

Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations
- PMID: 11416153
- PMCID: PMC87167
- DOI: 10.1128/MCB.21.14.4785-4806.2001
Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations
Abstract
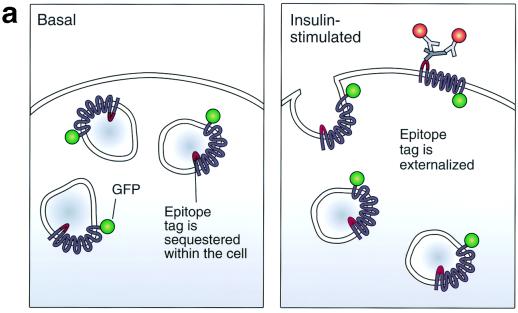
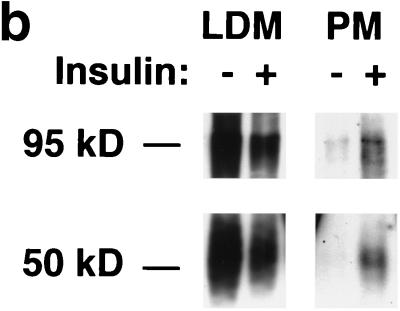
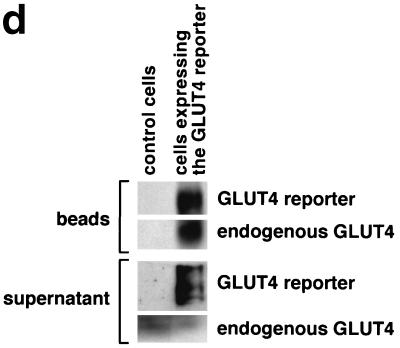
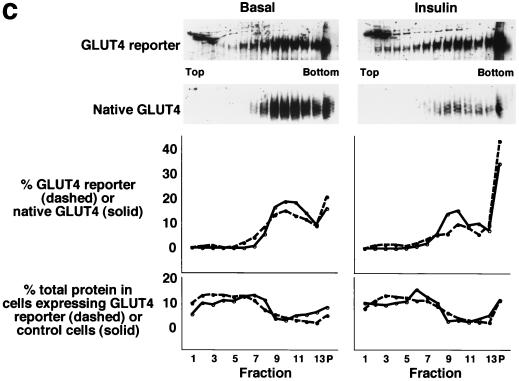
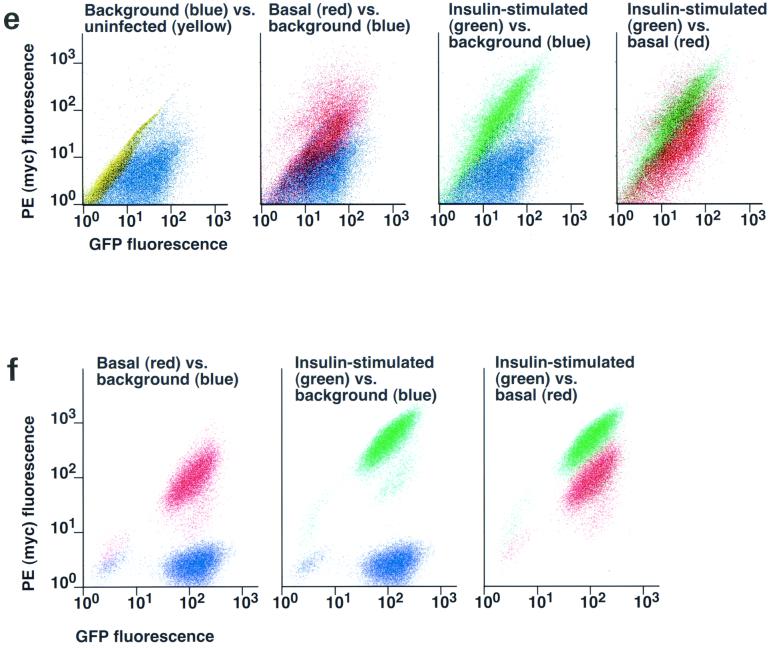
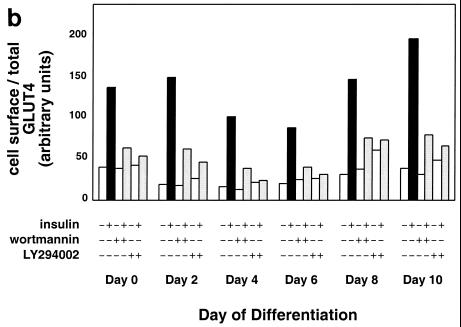
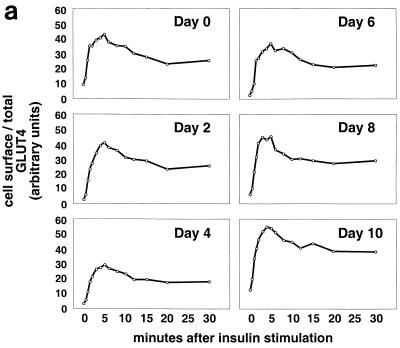
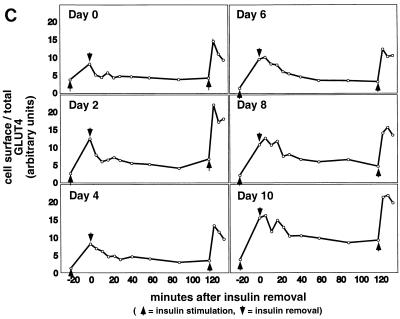
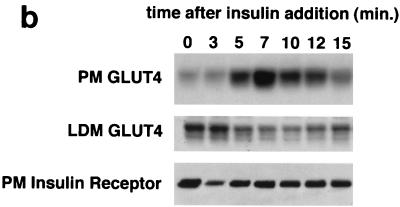
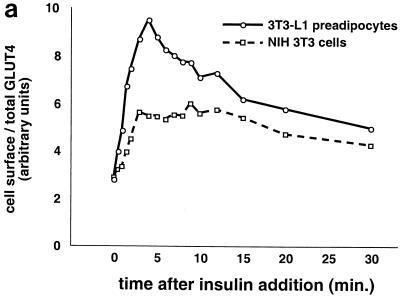
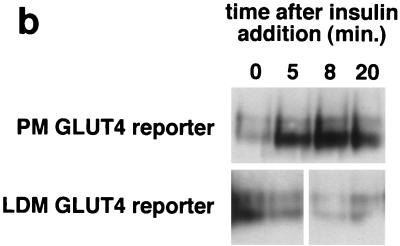
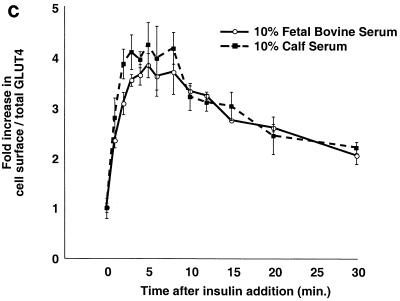
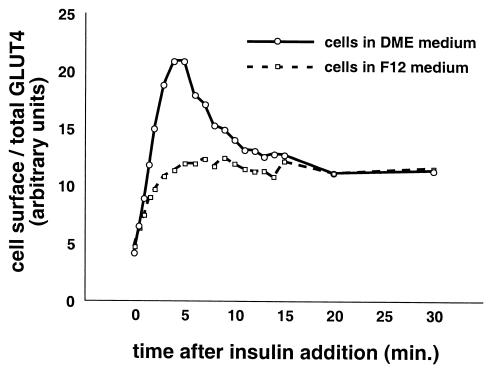
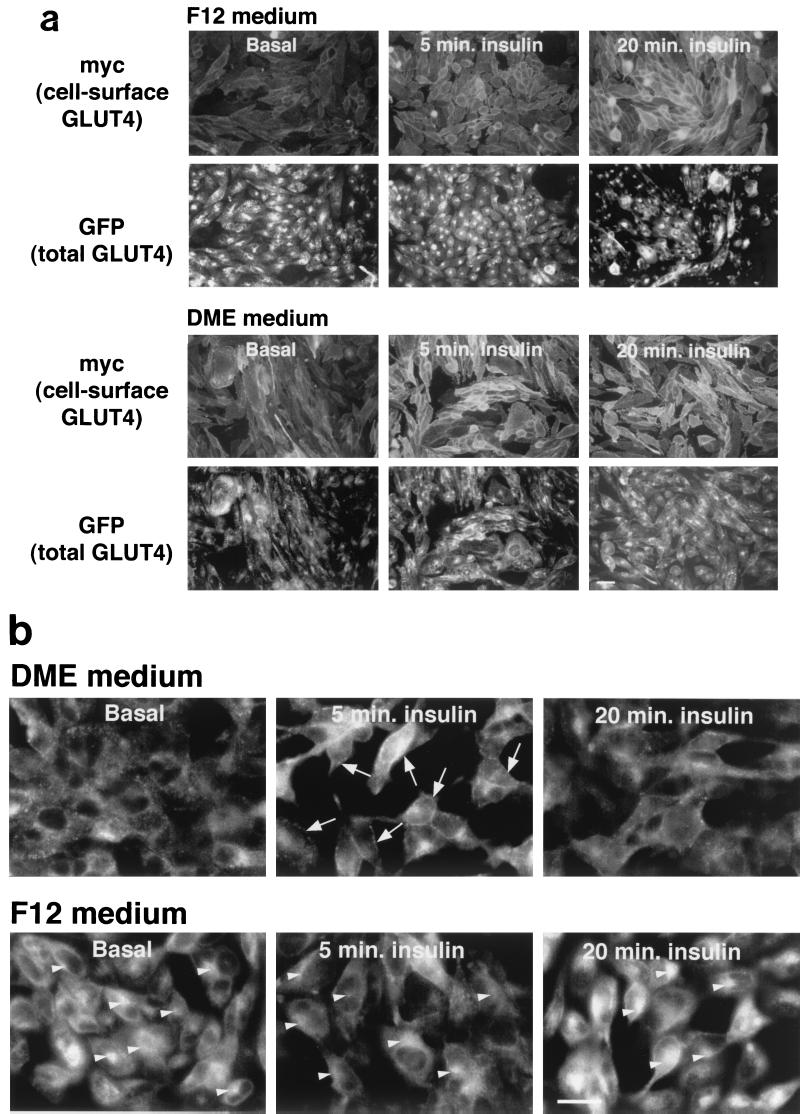
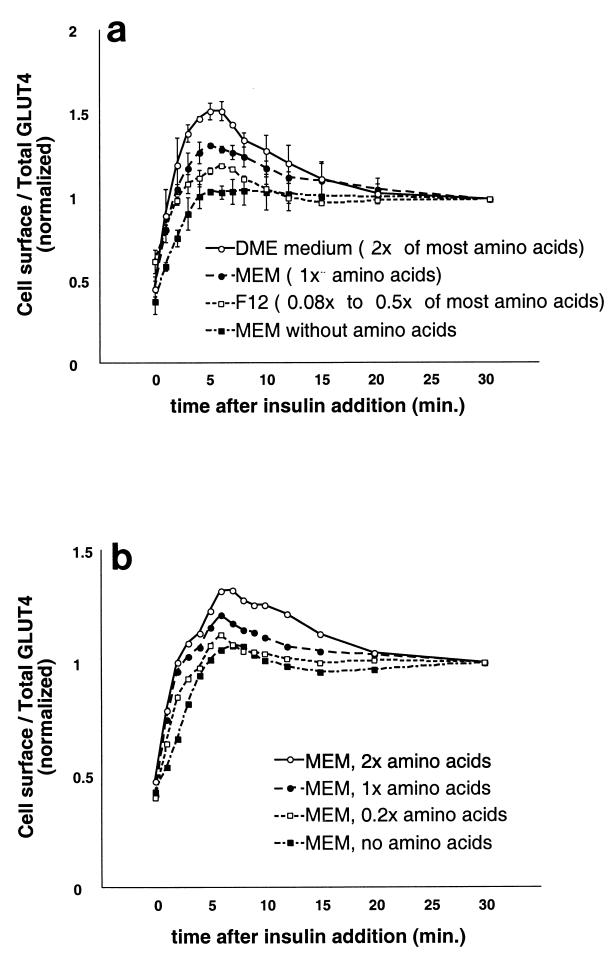
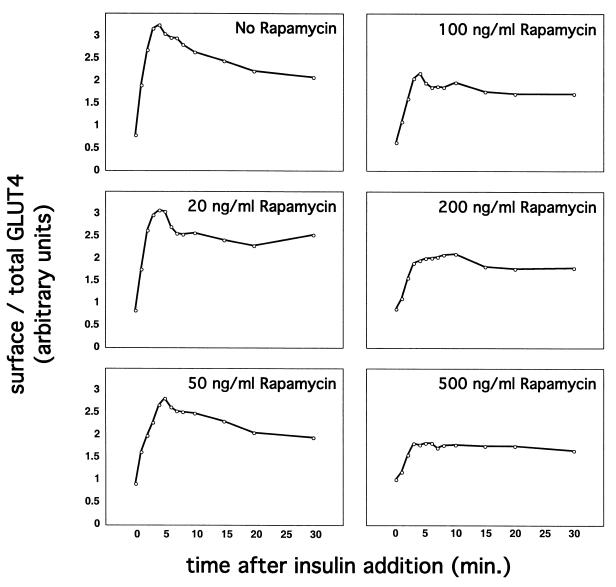
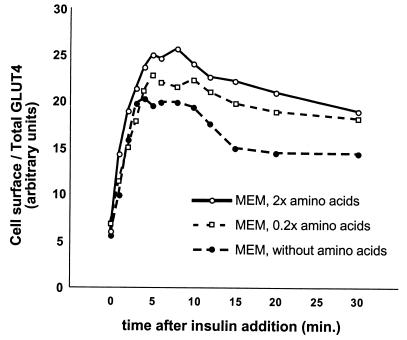
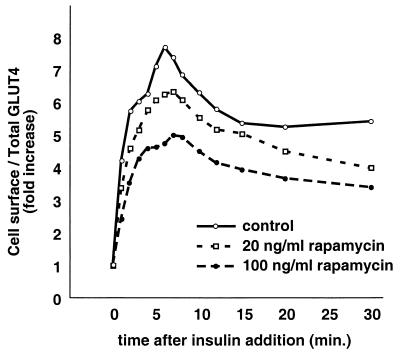
In fat and muscle, insulin stimulates glucose uptake by rapidly mobilizing the GLUT4 glucose transporter from a specialized intracellular compartment to the plasma membrane. We describe a method to quantify the relative proportion of GLUT4 at the plasma membrane, using flow cytometry to measure a ratio of fluorescence intensities corresponding to the cell surface and total amounts of a tagged GLUT4 reporter in individual living cells. Using this assay, we demonstrate that both 3T3-L1 and CHO cells contain intracellular compartments from which GLUT4 is rapidly mobilized by insulin and that the initial magnitude and kinetics of redistribution to the plasma membrane are similar in these two cell types when they are cultured identically. Targeting of GLUT4 to a highly insulin-responsive compartment in CHO cells is modulated by culture conditions. In particular, we find that amino acids regulate distribution of GLUT4 to this kinetically defined compartment through a rapamycin-sensitive pathway. Amino acids also modulate the magnitude of insulin-stimulated translocation in 3T3-L1 adipocytes. Our results indicate a novel link between glucose and amino acid metabolism.
Figures
References
-
- Al-Hasani H, Yver D R, Cushman S W. Overexpression of the glucose transporter GLUT4 in adipose cells interferes with insulin-stimulated translocation. FEBS Lett. 1999;460:338–342. - PubMed
-
- Asano T, Takata K, Katagiri H, Tsukuda K, Lin J L, Ishihara H, Inukai K, Hirano H, Yazaki Y, Oka Y. Domains responsible for the differential targeting of glucose transporter isoforms. J Biol Chem. 1992;267:19636–19641. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials