

Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome
- PMID: 11431708
- PMCID: PMC1235301
- DOI: 10.1086/321293
Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome
Abstract
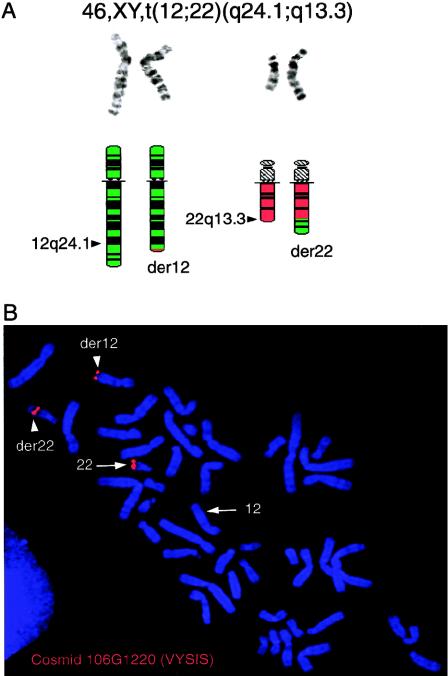
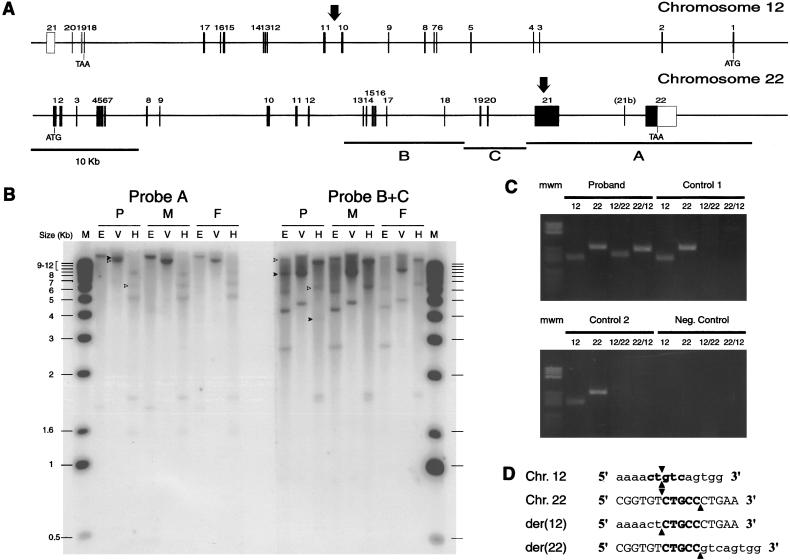
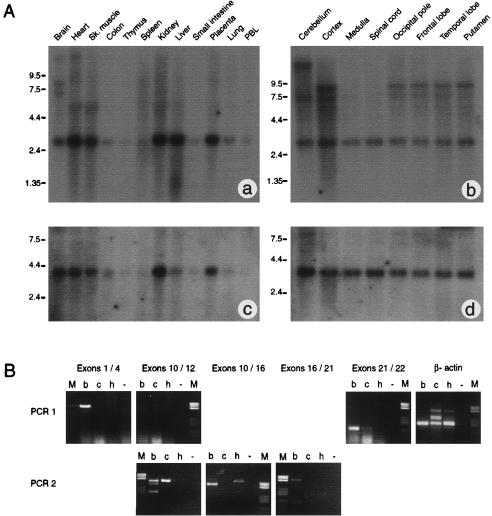
The terminal 22q13.3 deletion syndrome is characterized by severe expressive-language delay, mild mental retardation, hypotonia, joint laxity, dolichocephaly, and minor facial dysmorphisms. We identified a child with all the features of 22q13.3 deletion syndrome. The patient's karyotype showed a de novo balanced translocation between chromosomes 12 and 22, with the breakpoint in the 22q13.3 critical region of the 22q distal deletion syndrome [46, XY, t(12;22)(q24.1;q13.3)]. FISH investigations revealed that the translocation was reciprocal, with the chromosome 22 breakpoint within the 22q subtelomeric cosmid 106G1220 and the chromosome 12q breakpoint near STS D12S317. Using Southern blot analysis and inverse PCR, we located the chromosome 12 breakpoint in an intron of the FLJ10659 gene and located the chromosome 22 breakpoint within exon 21 of the human homologue of the ProSAP2 gene. Short homologous sequences (5-bp, CTG[C/A]C) were found at the breakpoint on both derivative chromosomes. The translocation does not lead to the loss of any portion of DNA. Northern blot analysis of human tissues, using the rat ProSAP2 cDNA, showed that full-length transcripts were found only in the cerebral cortex and the cerebellum. The FLJ10659 gene is expressed in various tissues and does not show tissue-specific isoforms. The finding that ProSAP2 is included in the critical region of the 22q deletion syndrome and that our proband displays all signs and symptoms of the syndrome suggests that ProSAP2 haploinsufficiency is the cause of the 22q13.3 deletion syndrome. ProSAP2 is a good candidate for this syndrome, because it is preferentially expressed in the cerebral cortex and the cerebellum and encodes a scaffold protein involved in the postsynaptic density of excitatory synapses.
Figures
References
Electronic-Database Information
-
- BLAST 2, http://www.ncbi.nlm.nih.gov/blast/blast.cgi (for BLAST analysis)
-
- Genome Database, http://www-genome.wi.mit.edu/cgi-bin/contig/phys_map (for locus FLJ10659)
-
- I.M.A.G.E. Consortium, http://image.llnl.gov/ (for I.M.A.G.E. clone 3450646)
-
- LocusLink, http://www.ncbi.nlm.nih.gov/LocusLink/ (for FLJ10659 gene structure)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for APPL [MIM 604299]) - PubMed
References
-
- Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla K-H, Sanmarti-Vila L, Wex H, Langnaese K, Bockmann J, Garner CC, Gundelfinger ED (1999a) Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J Neurosci 19:6506–6518 - PMC - PubMed
-
- Boeckers TM, Winter C, Smalla K-H, Kreutz MR, Bockmann J, Seidenbecher C, Garner CC, Gundelfinger ED (1999b) Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem Biophys Res Commun 264:247–252 - PubMed
-
- Chelly J (1999) Breakthroughs in molecular and cellular mechanisms underlying X-linked mental retardation. Hum Mol Genet 8:1833–1838 - PubMed
-
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, Dobyns WB, Ledbetter DH (1997 ) A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 6:147–155 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
