

SPINE: an integrated tracking database and data mining approach for identifying feasible targets in high-throughput structural proteomics
- PMID: 11433035
- PMCID: PMC55760
- DOI: 10.1093/nar/29.13.2884
SPINE: an integrated tracking database and data mining approach for identifying feasible targets in high-throughput structural proteomics
Abstract
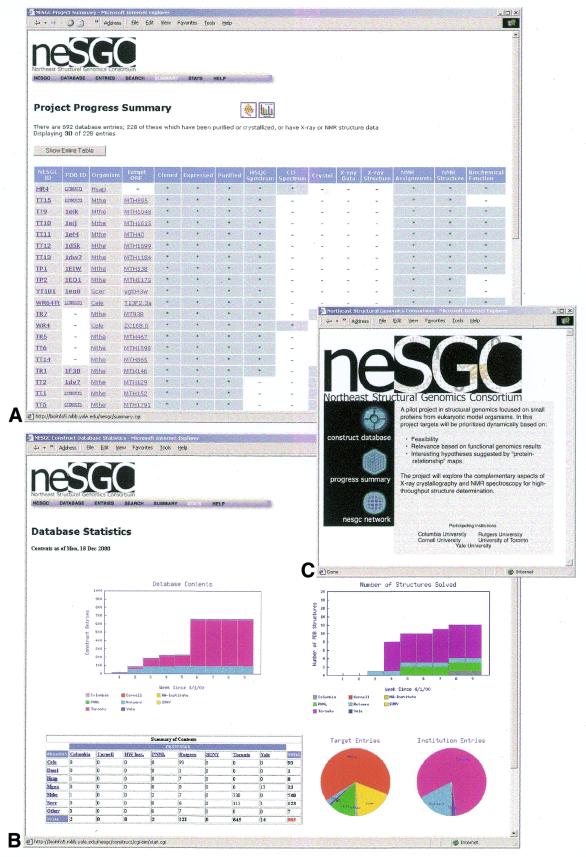
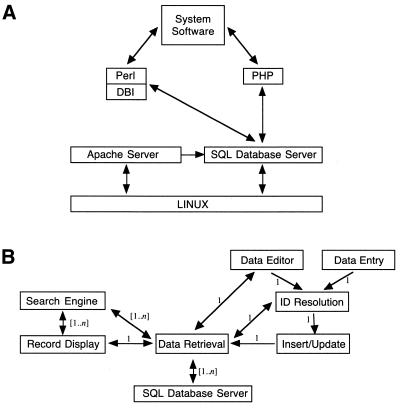
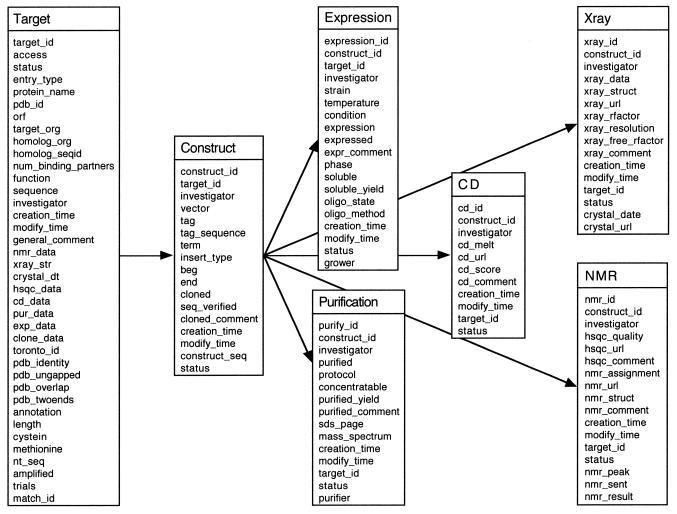
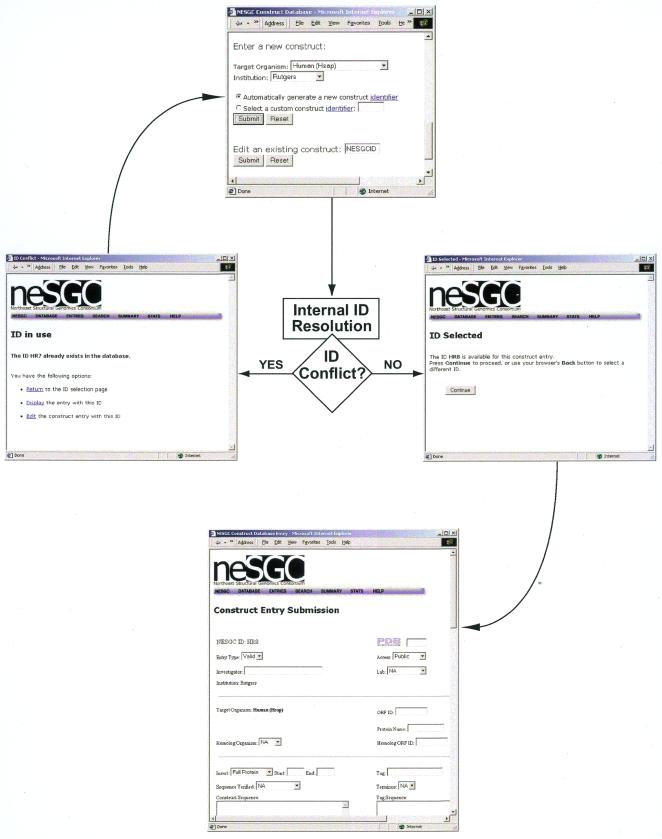
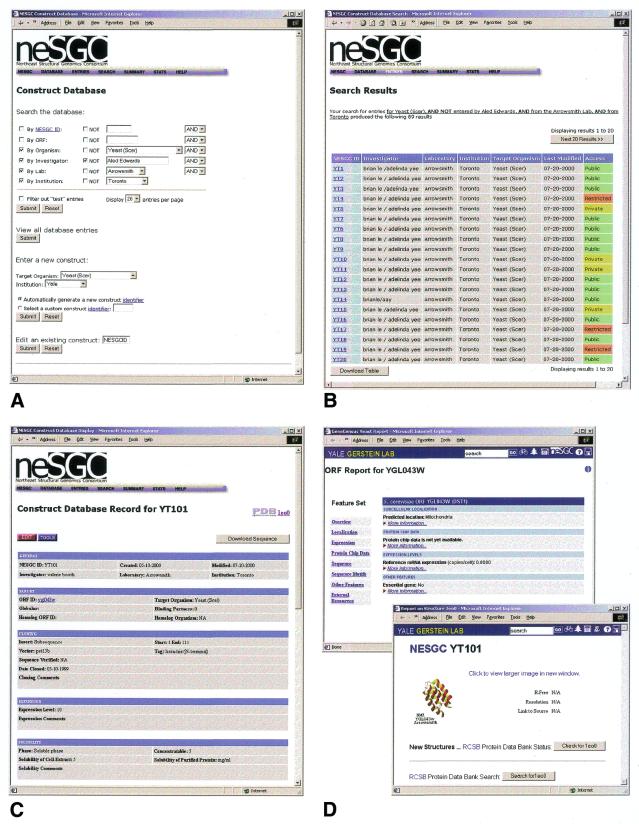
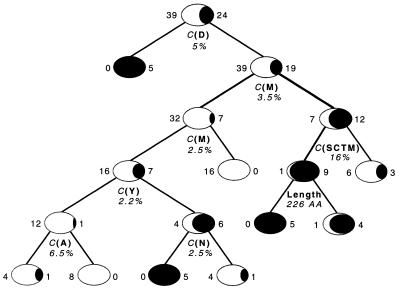
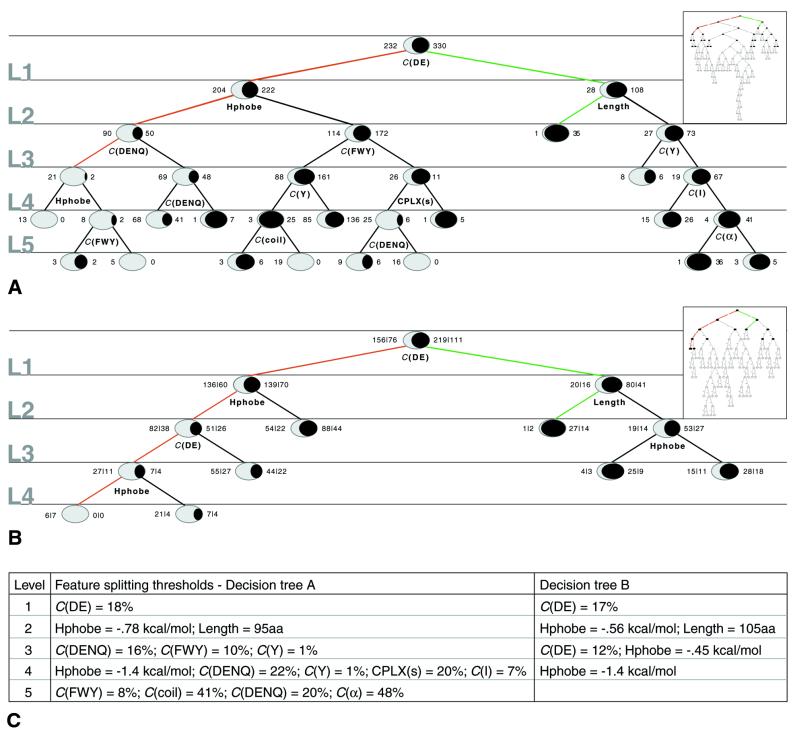
High-throughput structural proteomics is expected to generate considerable amounts of data on the progress of structure determination for many proteins. For each protein this includes information about cloning, expression, purification, biophysical characterization and structure determination via NMR spectroscopy or X-ray crystallography. It will be essential to develop specifications and ontologies for standardizing this information to make it amenable to retrospective analysis. To this end we created the SPINE database and analysis system for the Northeast Structural Genomics Consortium. SPINE, which is available at bioinfo.mbb.yale.edu/nesg or nesg.org, is specifically designed to enable distributed scientific collaboration via the Internet. It was designed not just as an information repository but as an active vehicle to standardize proteomics data in a form that would enable systematic data mining. The system features an intuitive user interface for interactive retrieval and modification of expression construct data, query forms designed to track global project progress and external links to many other resources. Currently the database contains experimental data on 985 constructs, of which 740 are drawn from Methanobacterium thermoautotrophicum, 123 from Saccharomyces cerevisiae, 93 from Caenorhabditis elegans and the remainder from other organisms. We developed a comprehensive set of data mining features for each protein, including several related to experimental progress (e.g. expression level, solubility and crystallization) and 42 based on the underlying protein sequence (e.g. amino acid composition, secondary structure and occurrence of low complexity regions). We demonstrate in detail the application of a particular machine learning approach, decision trees, to the tasks of predicting a protein's solubility and propensity to crystallize based on sequence features. We are able to extract a number of key rules from our trees, in particular that soluble proteins tend to have significantly more acidic residues and fewer hydrophobic stretches than insoluble ones. One of the characteristics of proteomics data sets, currently and in the foreseeable future, is their intermediate size ( approximately 500-5000 data points). This creates a number of issues in relation to error estimation. Initially we estimate the overall error in our trees based on standard cross-validation. However, this leaves out a significant fraction of the data in model construction and does not give error estimates on individual rules. Therefore, we present alternative methods to estimate the error in particular rules.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
