

Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine
- PMID: 11438586
- PMCID: PMC6762835
- DOI: 10.1523/JNEUROSCI.21-14-05110.2001
Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine
Abstract
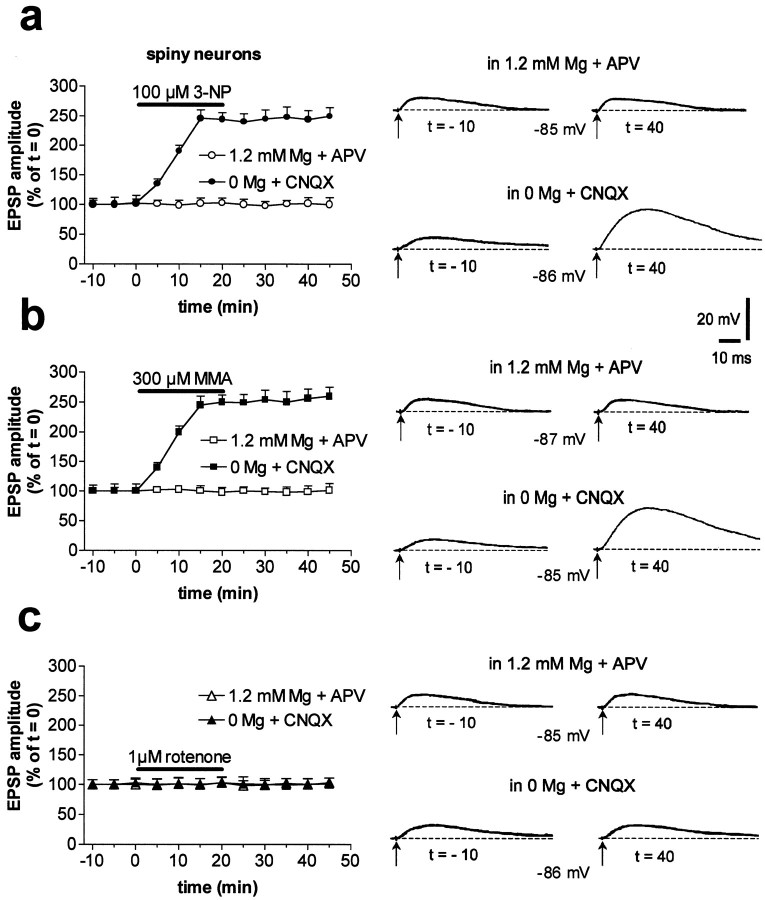
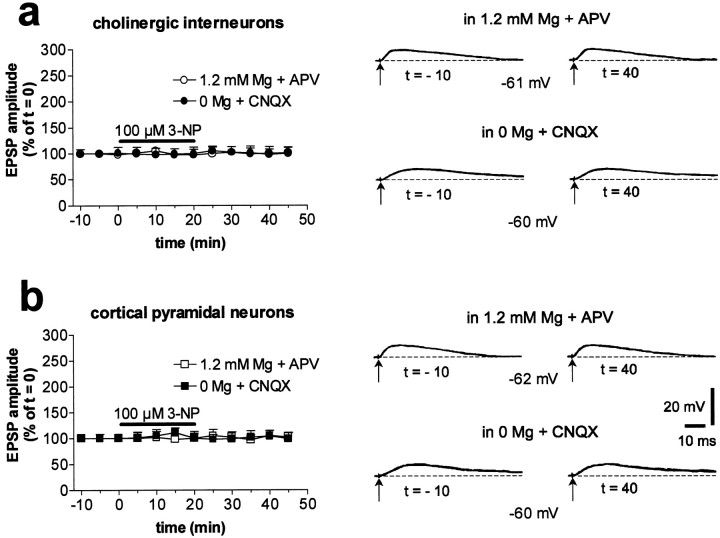
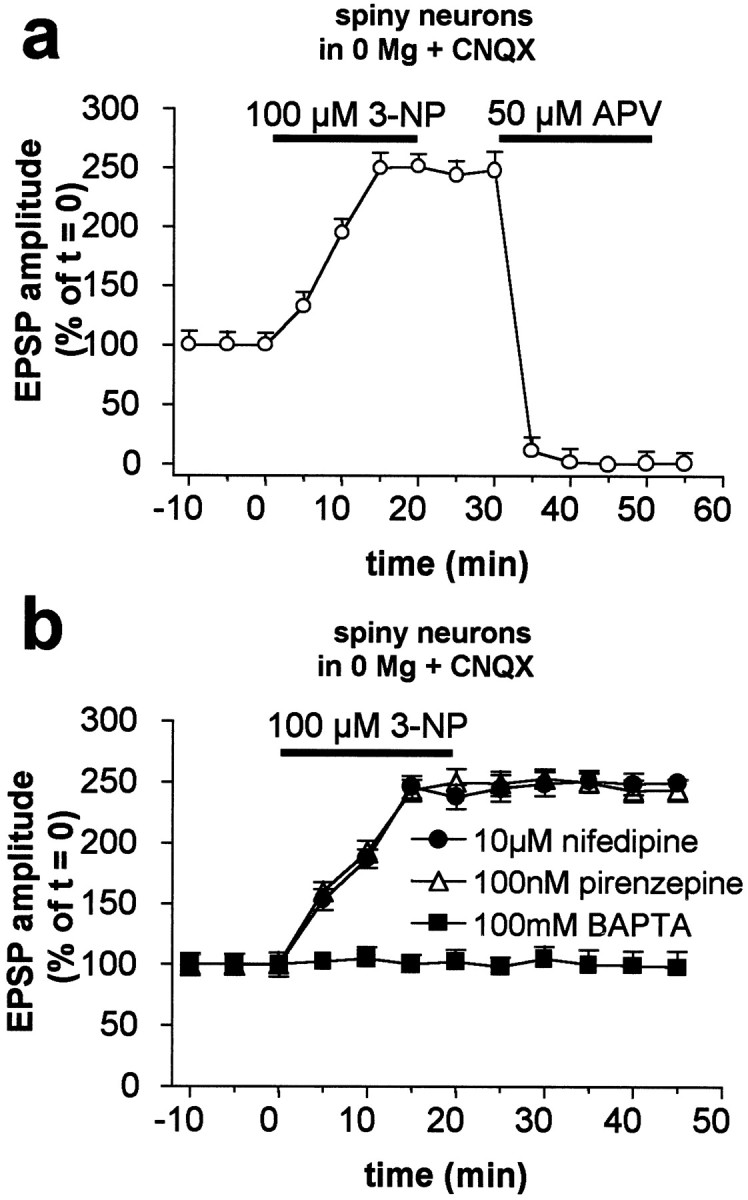
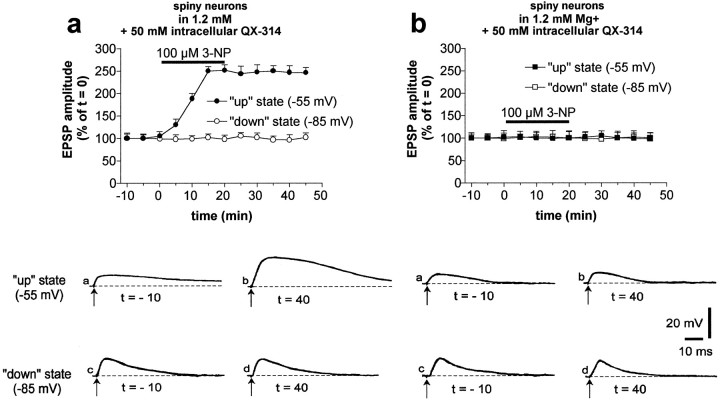
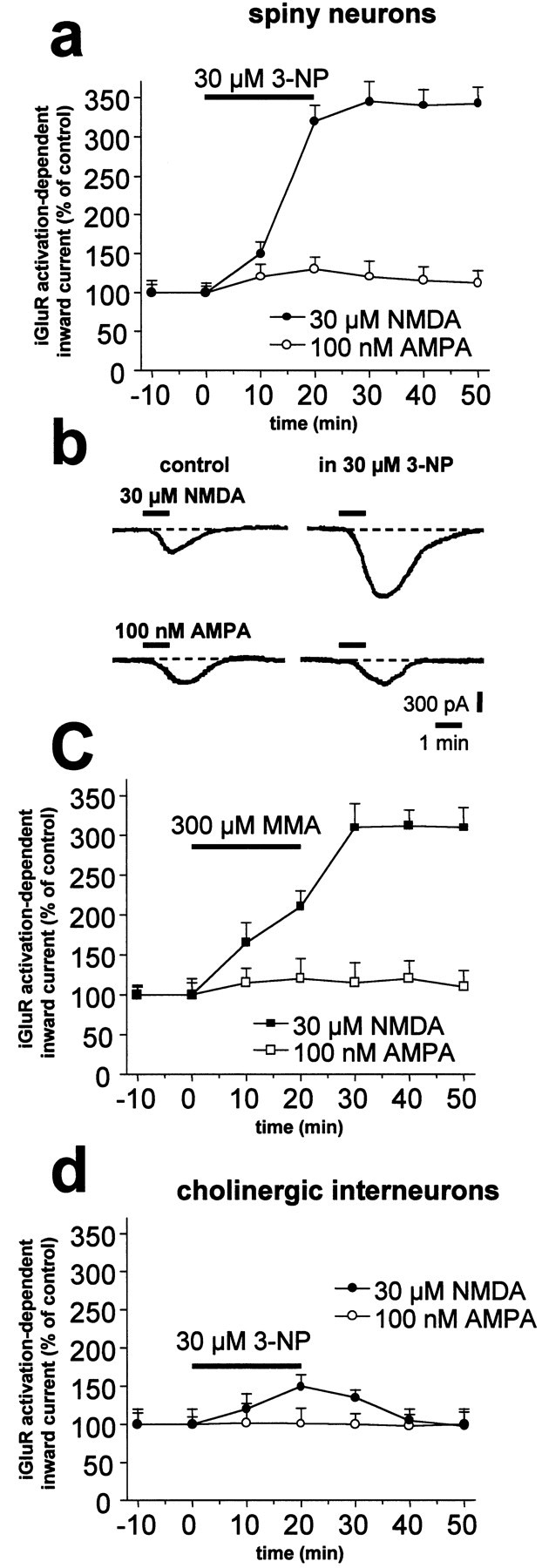
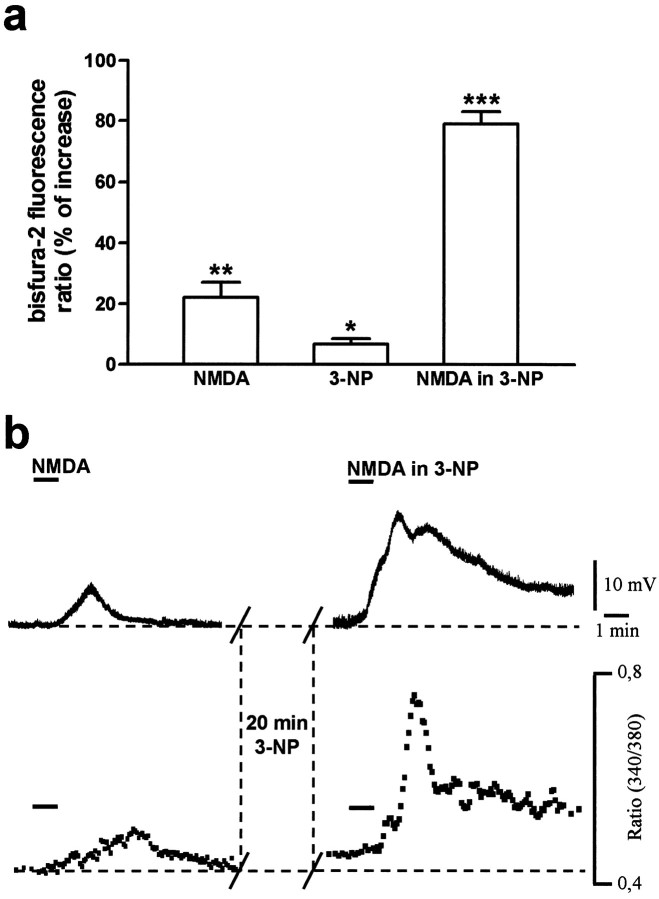
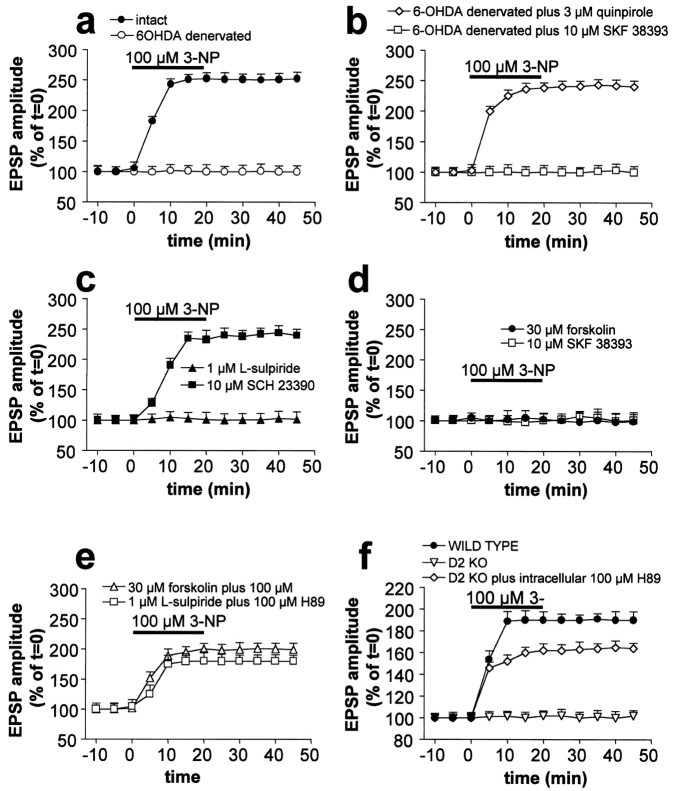
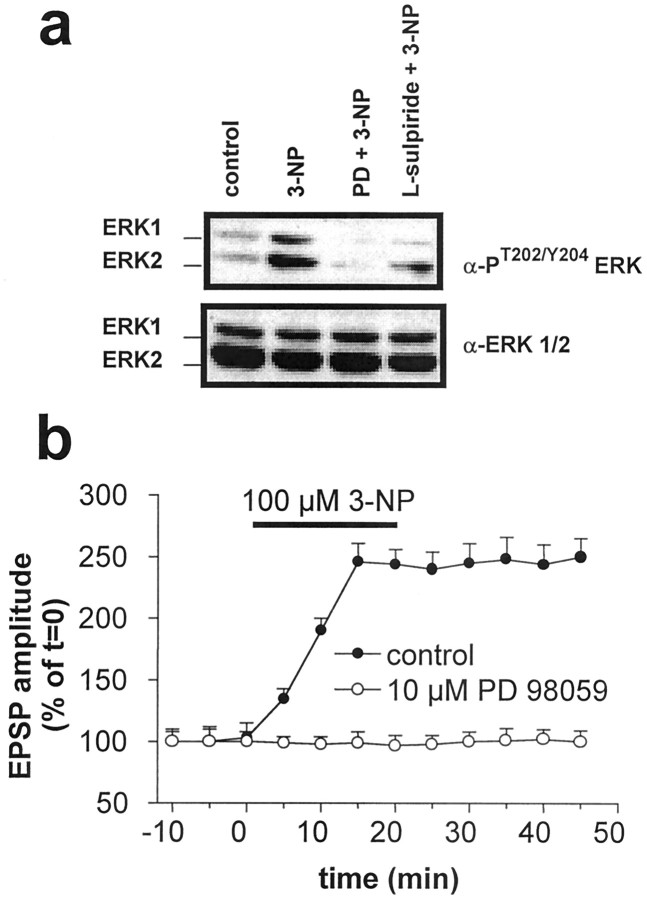
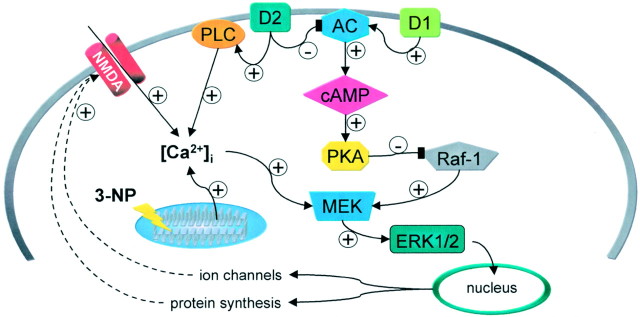
Abnormal involuntary movements and cognitive impairment represent the classical clinical symptoms of Huntington's disease (HD). This genetic disorder involves degeneration of striatal spiny neurons, but not striatal large cholinergic interneurons, and corresponds to a marked decrease in the activity of mitochondrial complex II [succinate dehydrogenase (SD)] in the brains of HD patients. Here we have examined the possibility that SD inhibitors exert their toxic action by increasing glutamatergic transmission. We report that SD inhibitors such as 3-nitroproprionic acid (3-NP), but not an inhibitor of mitochondrial complex I, produce a long-term potentiation of the NMDA-mediated synaptic excitation (3-NP-LTP) in striatal spiny neurons. In contrast, these inhibitors had no effect on excitatory synaptic transmission in striatal cholinergic interneurons and pyramidal cortical neurons. 3-NP-LTP involves increased intracellular calcium and activation of the mitogen-activated protein kinase extracellular signal-regulated kinase and is critically dependent on endogenous dopamine acting via D2 receptors, whereas it is negatively regulated by D1 receptors. Thus 3-NP-LTP might play a key role in the regional and cell type-specific neuronal death observed in HD.
Figures
References
-
- Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–230. - PubMed
-
- Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington's disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998;43:253–255. - PubMed
-
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice like dopamine D2 receptors. Nature. 1995;377:424–428. - PubMed
-
- Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem. 1996;67:593–600. - PubMed
-
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous