

c-Myc is necessary for DNA damage-induced apoptosis in the G(2) phase of the cell cycle
- PMID: 11438650
- PMCID: PMC87219
- DOI: 10.1128/MCB.21.15.4929-4937.2001
c-Myc is necessary for DNA damage-induced apoptosis in the G(2) phase of the cell cycle
Abstract
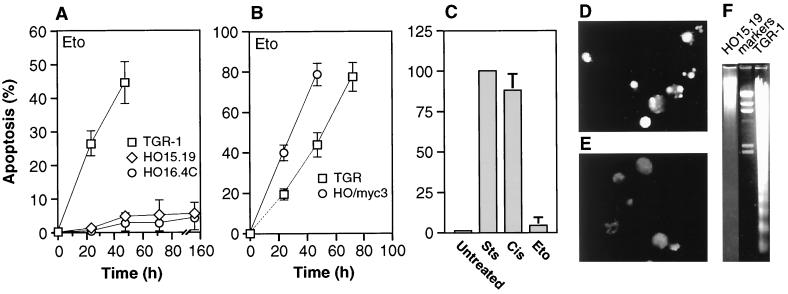
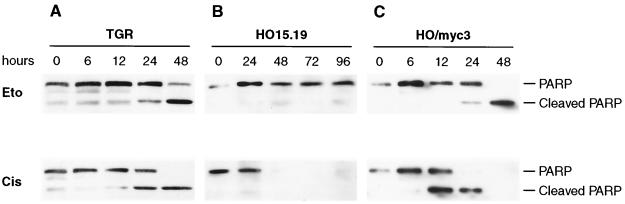
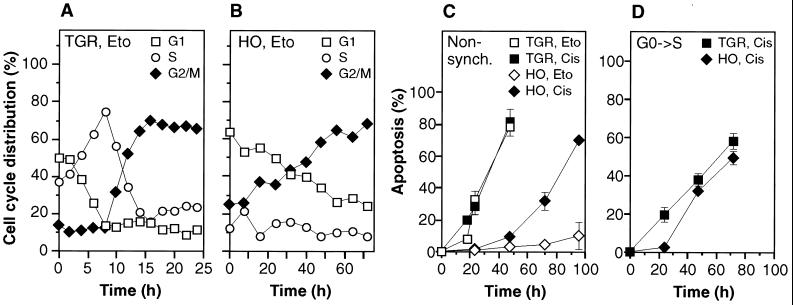
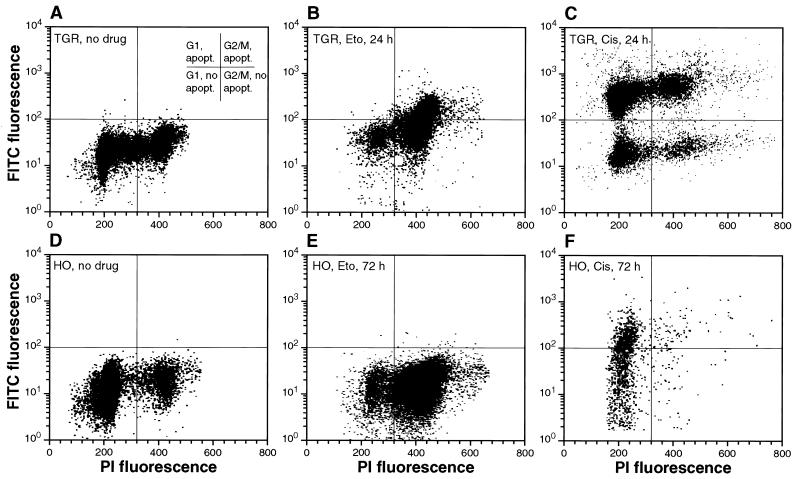
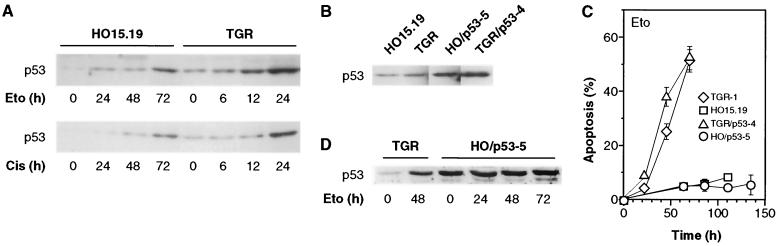
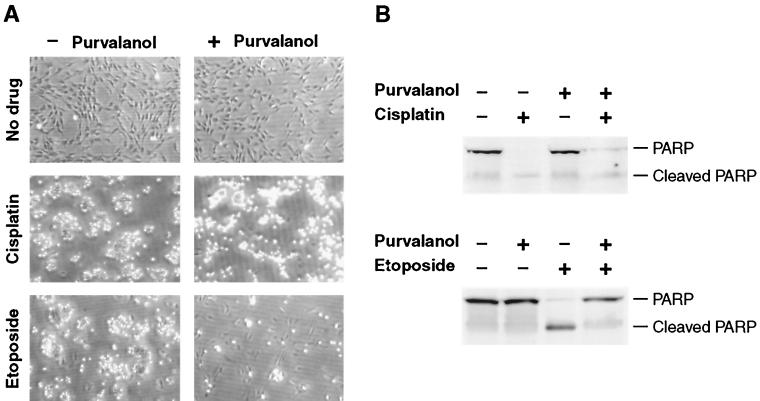
The c-myc proto-oncogene encodes a transcription factor that participates in the regulation of cellular proliferation, differentiation, and apoptosis. Ectopic overexpression of c-Myc has been shown to sensitize cells to apoptosis. We report here that cells lacking c-Myc activity due to disruption of the c-myc gene by targeted homologous recombination are defective in DNA damage-initiated apoptosis in the G(2) phase of the cell cycle. The downstream effector of c-Myc is cyclin A, whose ectopic expression in c-myc(-/-) cells rescues the apoptosis defect. The kinetics of the G(2) response indicate that the induction of cyclin A and the concomitant activation of Cdk2 represent an early step during commitment to apoptosis. In contrast, expression of cyclins E and D1 does not rescue the apoptosis defect, and apoptotic processes in G(1) phase are not affected in c-myc(-/-) cells. These observations link DNA damage-induced apoptosis with cell cycle progression and implicate c-Myc in the functioning of a subset of these pathways.
Figures

References
-
- Adachi S, Ito H, Tamamori-Adachi M, Ono Y, Nozato T, Abe S, Ikeda M, Marumo F, Hiroe M. Cyclin A/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes. Circ Res. 2001;88:408–414. - PubMed
-
- Askew D S, Ashmun R A, Simmons B C, Cleveland J L. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. - PubMed
-
- Canelles M, Delgado M D, Hyland K M, Lerga A, Richard C, Dang C V, Leon J. Max and inhibitory c-Myc mutants induce erythroid differentiation and resistance to apoptosis in human myeloid leukemia calls. Oncogene. 1997;14:1315–1327. - PubMed
-
- Chang Y T, Gray N S, Rosania G R, Sutherlin D P, Kwon S, Norman T C, Sarohia R, Leost M, Meijer L, Schultz P G. Synthesis and application of functionally diverse 2,6,9-trisubstituted purine libraries as CDK inhibitors. Chem Biol. 1999;6:361–375. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources