

Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation
- PMID: 11457894
- PMCID: PMC2193459
- DOI: 10.1084/jem.194.2.189
Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation
Abstract
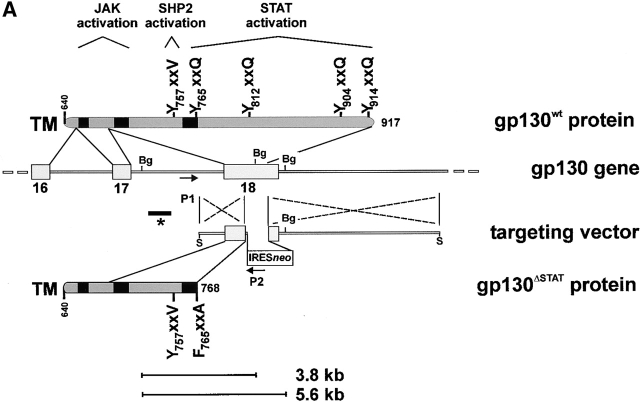
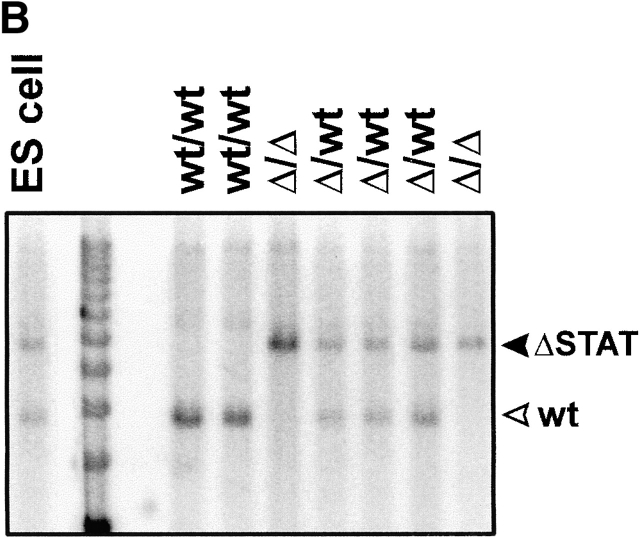
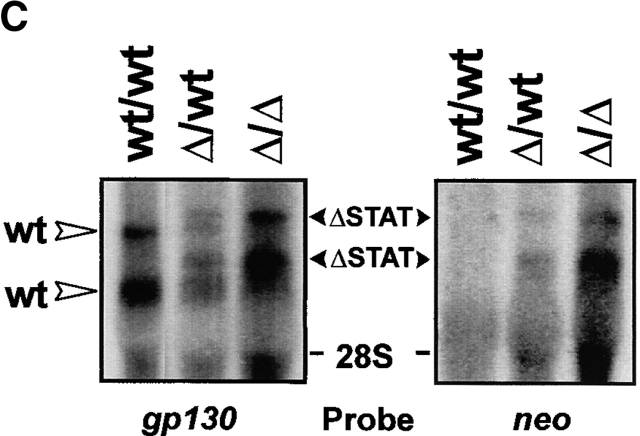
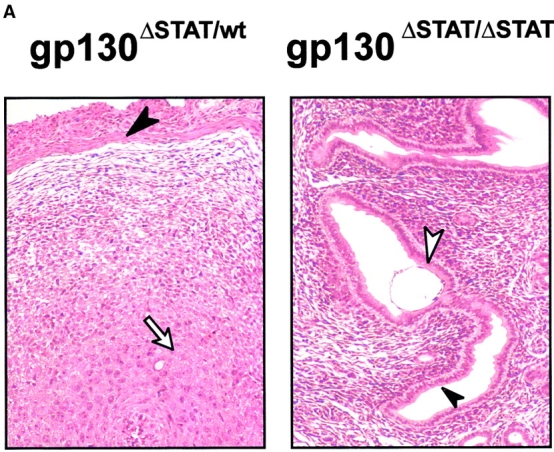
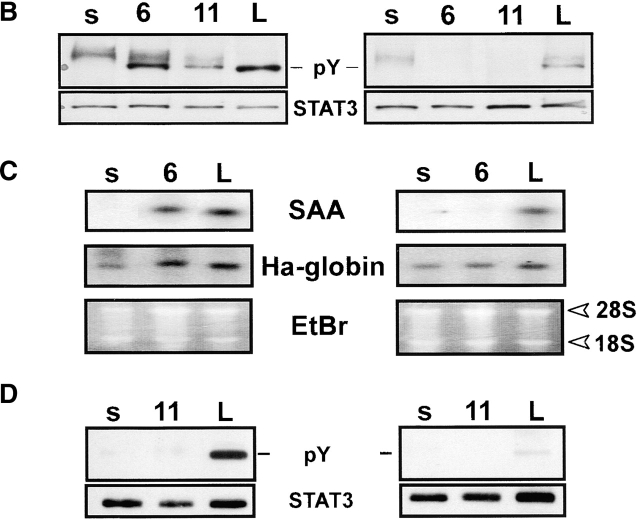
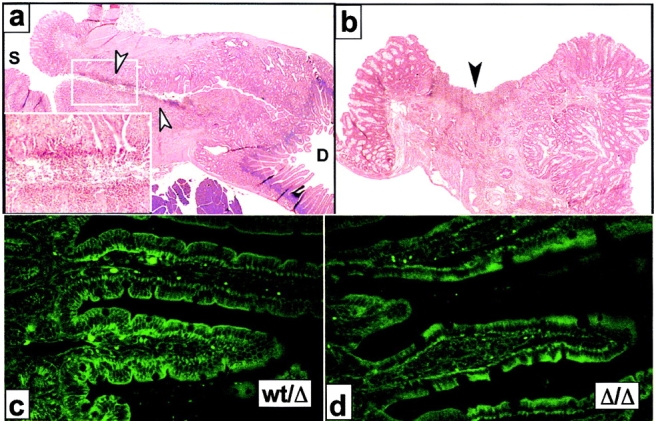
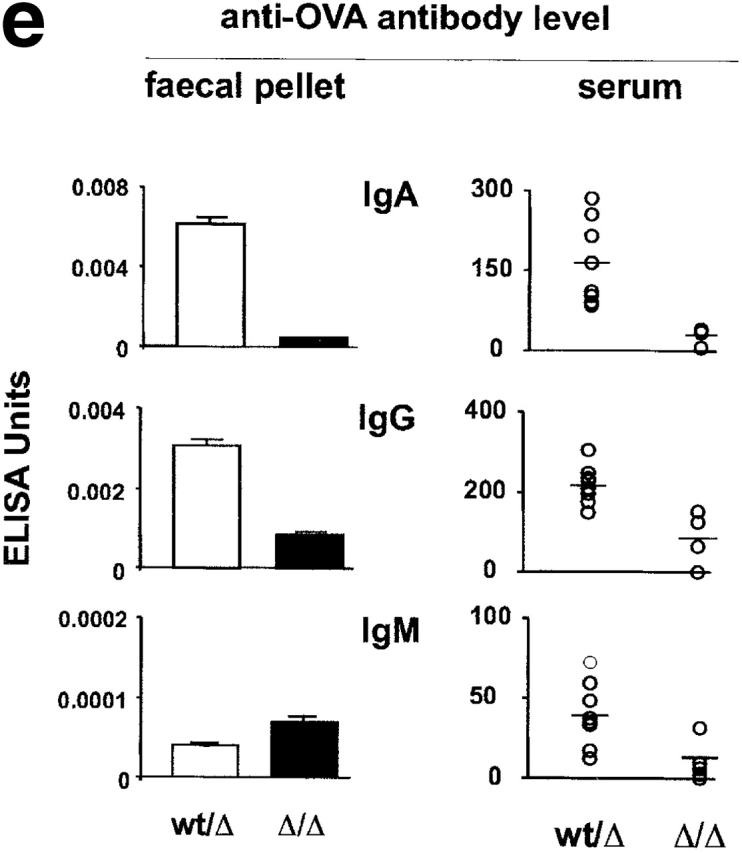
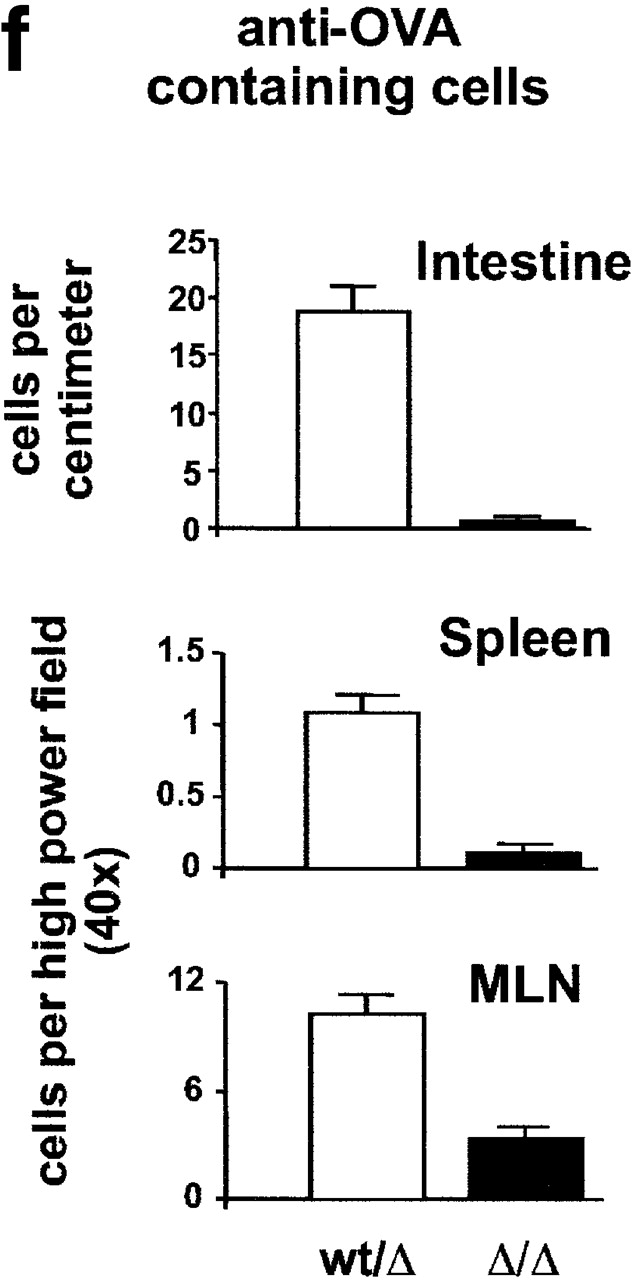
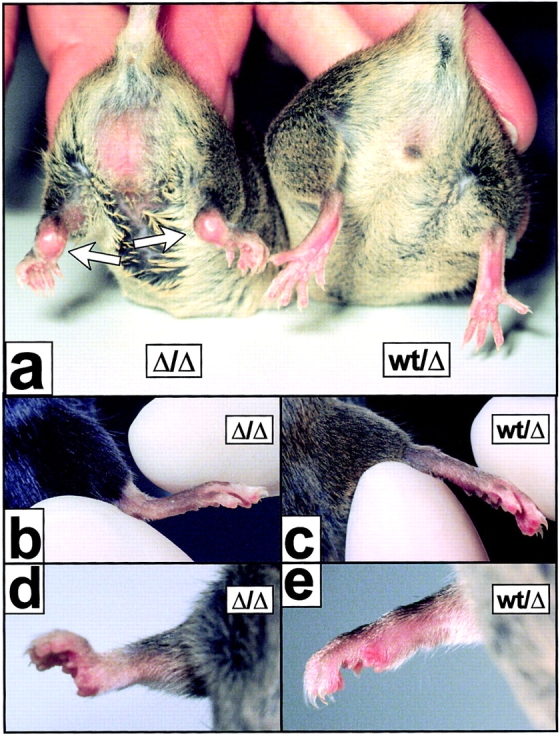
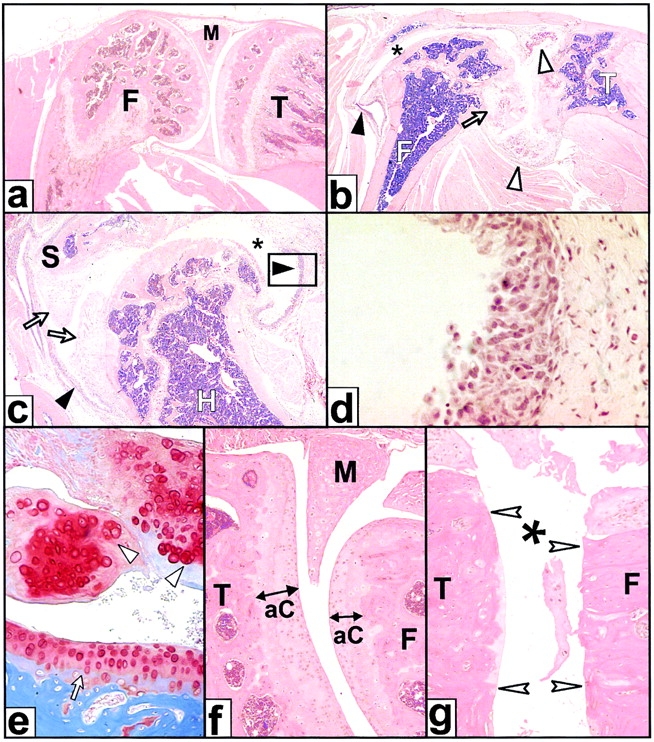
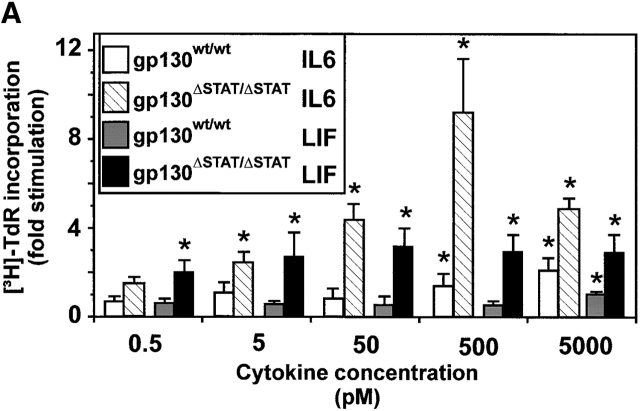
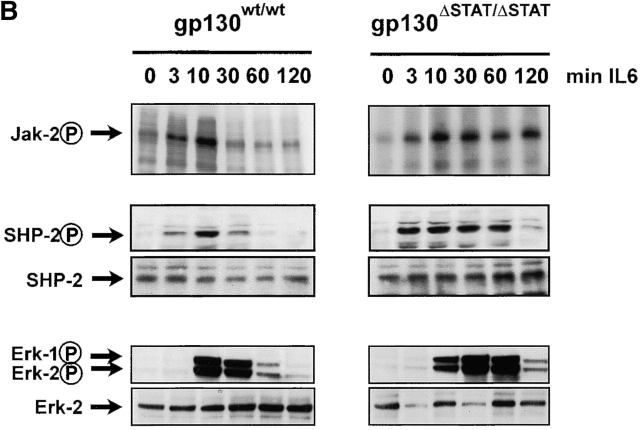
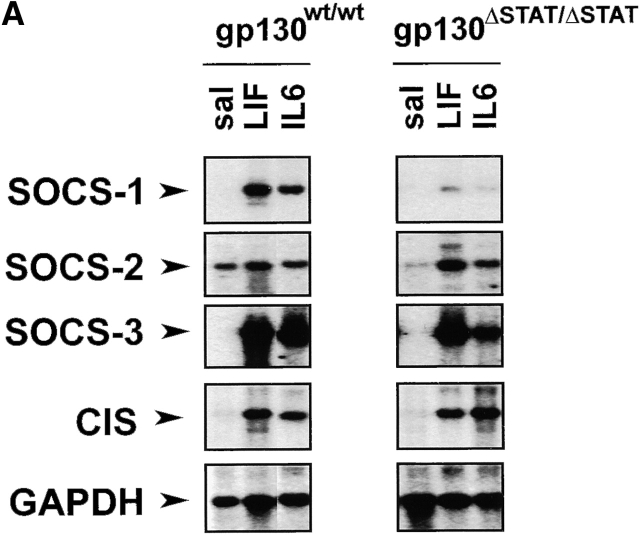
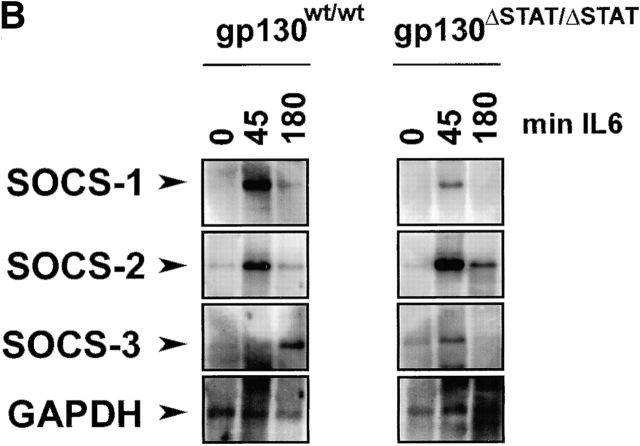
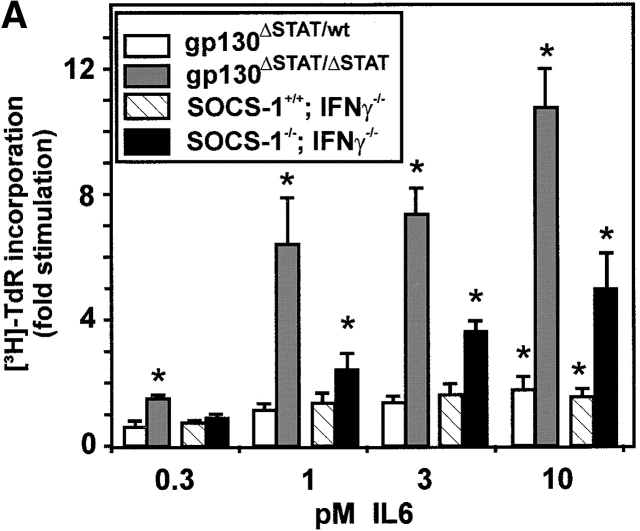
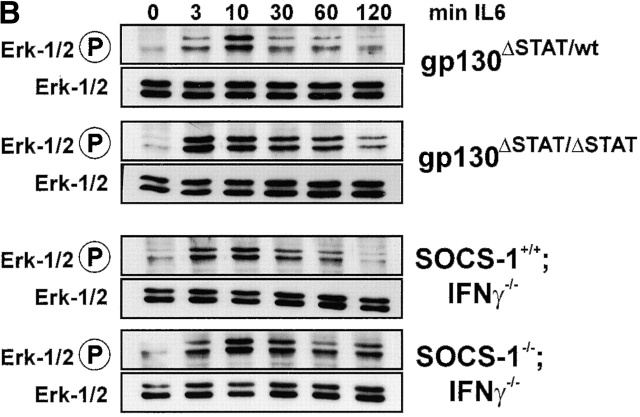
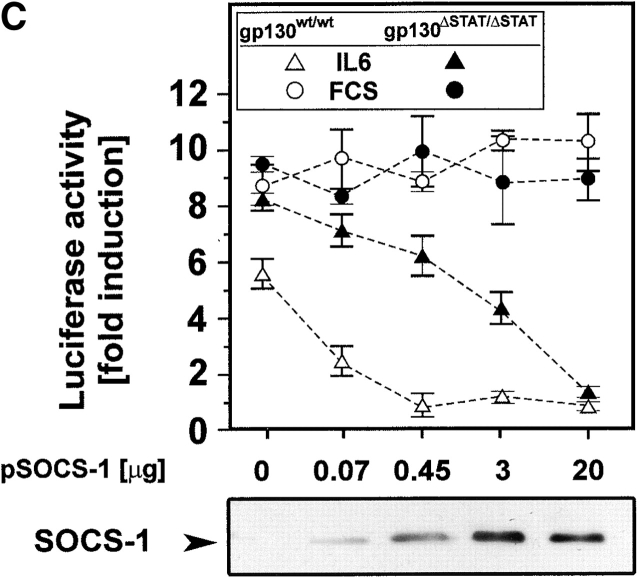
The receptor subunit gp130 transduces multiple cell type-specific activities of the leukemia inhibitory factor (LIF)/interleukin (IL)-6 family of cytokines through the signal transducer and activator of transcription (STAT) and src homology 2 domain-bearing protein tyrosine phosphatase (SHP)-2/ras/Erk pathways. To define STAT-dependent physiological responses, we generated mice with a COOH-terminal gp130(DeltaSTAT) "knock-in" mutation which deleted all STAT-binding sites. gp130(DeltaSTAT) mice phenocopyed mice deficient for IL-6 (impaired humoral and mucosal immune and hepatic acute phase responses) and LIF (failure of blastocyst implantation). However, unlike mice with null mutations in any of the components in the gp130 signaling pathway, gp130(DeltaSTAT) mice also displayed gastrointestinal ulceration and a severe joint disease with features of chronic synovitis, cartilaginous metaplasia, and degradation of the articular cartilage. Mitogenic hyperresponsiveness of synovial cells to the LIF/IL-6 family of cyto-kines was caused by sustained gp130-mediated SHP-2/ras/Erk activation due to impaired STAT-mediated induction of suppressor of cytokine signaling (SOCS) proteins which normally limits gp130 signaling. Therefore, the joint pathology in gp130(DeltaSTAT) mice is likely to arise from the disturbance of the otherwise balanced activation of the SHP-2/ras/Erk and STAT signaling cascades emanating from gp130.
Figures
References
-
- Taga T., Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997;15:797–819. - PubMed
-
- Waring P. Leukemia inhibitory factor. In: Garland J.M., editor. Colony Stimulating Factors, Molecular and Cellular Biology. Second ed. Marcel Dekker Publishers; New York: 1997. pp. 467–513.
-
- Kopf M., Baumann H., Freer G., Freudenberg M., Lamers M., Kishimoto T., Zinkernagel R., Bluethmann H., Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. - PubMed
-
- Ramsay A.J., Husband A.J., Ramshaw I.A., Bao S., Matthaei K.I., Koehler G., Kopf M. The role of interleukin-6 in mucosal IgA antibody responses in vivo. Science. 1994;264:561–563. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
