

Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum
- PMID: 11466286
- PMCID: PMC99537
- DOI: 10.1128/JB.183.16.4823-4838.2001
Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum
Abstract
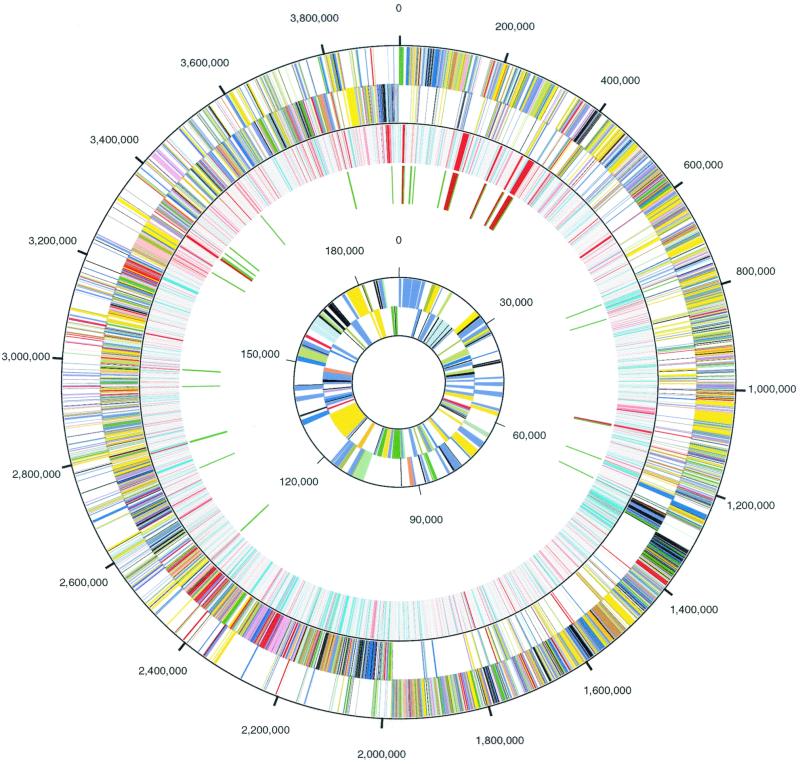
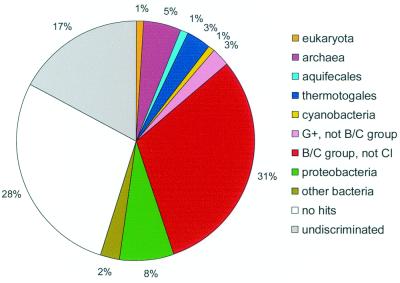
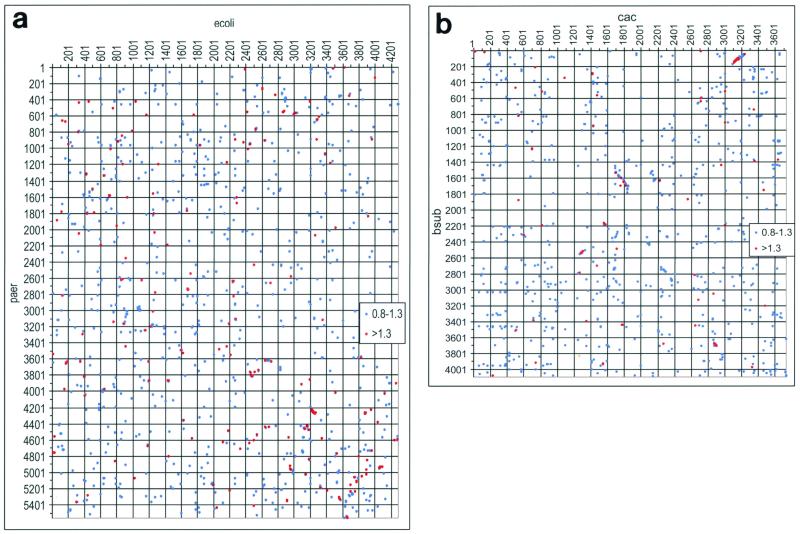
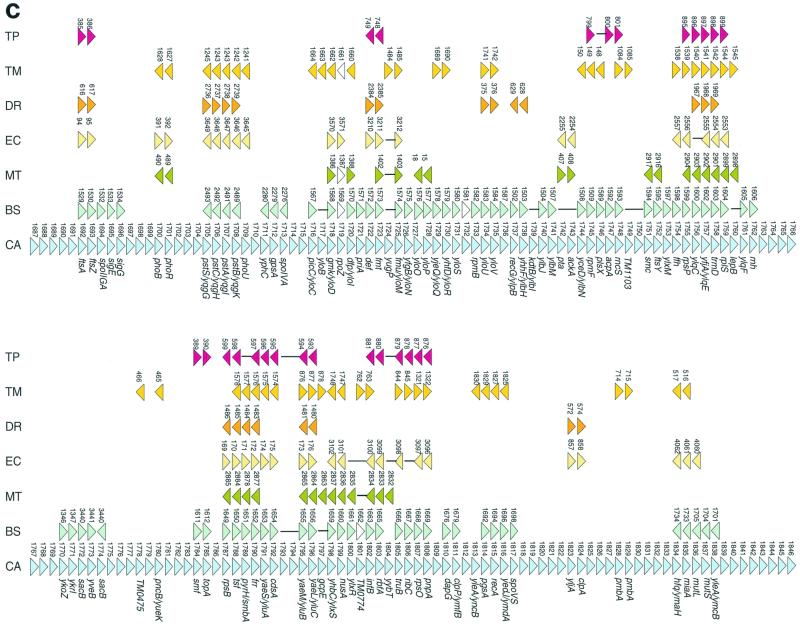
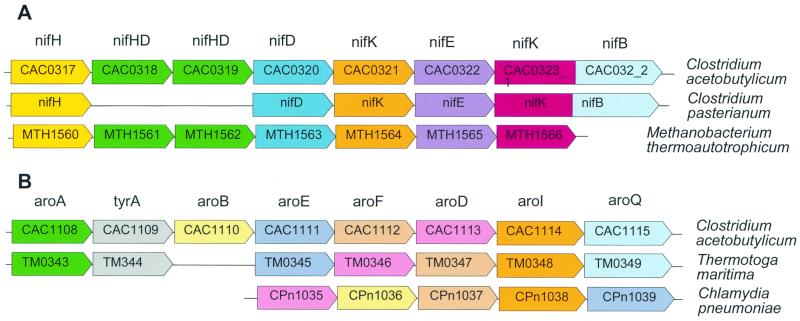
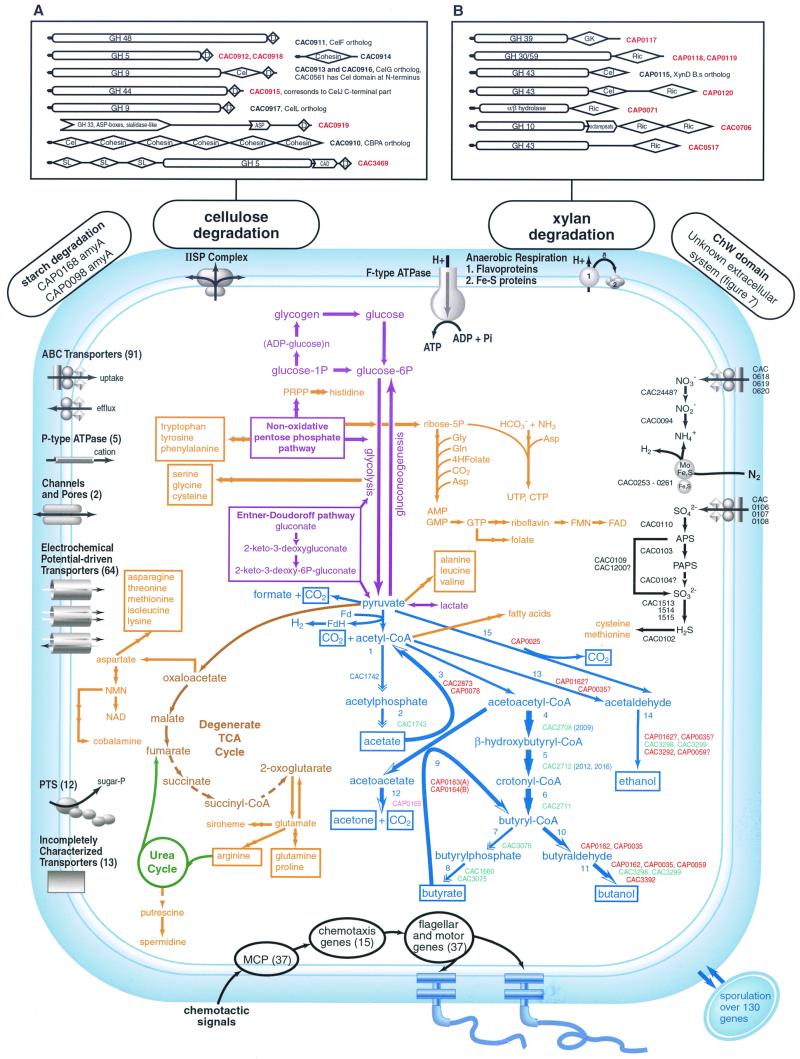
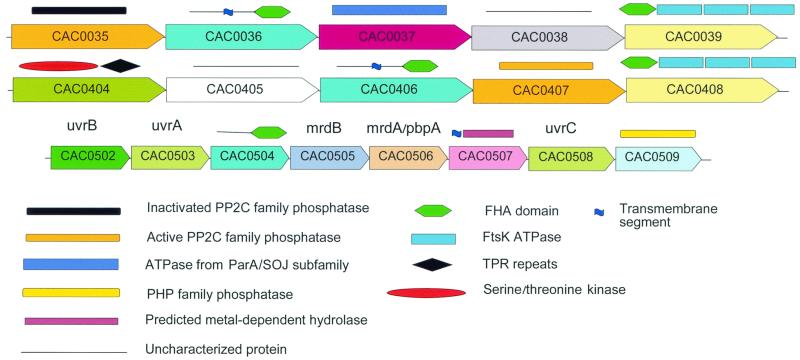
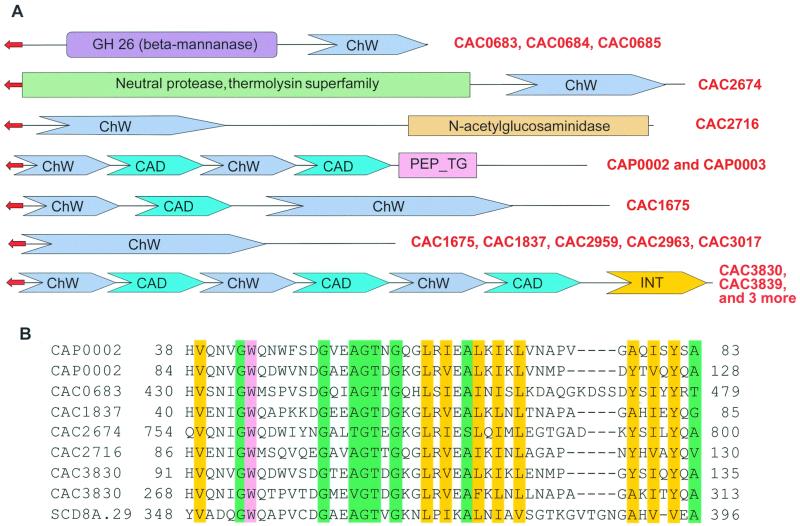
The genome sequence of the solvent-producing bacterium Clostridium acetobutylicum ATCC 824 has been determined by the shotgun approach. The genome consists of a 3.94-Mb chromosome and a 192-kb megaplasmid that contains the majority of genes responsible for solvent production. Comparison of C. acetobutylicum to Bacillus subtilis reveals significant local conservation of gene order, which has not been seen in comparisons of other genomes with similar, or, in some cases closer, phylogenetic proximity. This conservation allows the prediction of many previously undetected operons in both bacteria. However, the C. acetobutylicum genome also contains a significant number of predicted operons that are shared with distantly related bacteria and archaea but not with B. subtilis. Phylogenetic analysis is compatible with the dissemination of such operons by horizontal transfer. The enzymes of the solventogenesis pathway and of the cellulosome of C. acetobutylicum comprise a new set of metabolic capacities not previously represented in the collection of complete genomes. These enzymes show a complex pattern of evolutionary affinities, emphasizing the role of lateral gene exchange in the evolution of the unique metabolic profile of the bacterium. Many of the sporulation genes identified in B. subtilis are missing in C. acetobutylicum, which suggests major differences in the sporulation process. Thus, comparative analysis reveals both significant conservation of the genome organization and pronounced differences in many systems that reflect unique adaptive strategies of the two gram-positive bacteria.
Figures
References
-
- Altschul S F, Koonin E V. Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem Sci. 1998;23:444–447. - PubMed
-
- Bahl H, Mueller H, Behrens S, Joseph H, Narberhaus F. Expression of heat shock genes in Clostridium acetobutylicum. FEMS Microbiol Rev. 1995;17:341–348. - PubMed
-
- Bayer E A, Shimon L J, Shoham Y, Lamed R. Cellulosomes—structure and ultrastructure. J Struct Biol. 1998;124:221–234. - PubMed
-
- Blanchet, D., R. Marchal, and J. P. Vandecasteele. 1985. Acetone and butanol by fermentation of inulin. French patent 2559160.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
