

Modeling of the bacterial luciferase-flavin mononucleotide complex combining flexible docking with structure-activity data
- PMID: 11468353
- PMCID: PMC2374094
- DOI: 10.1110/ps.7201
Modeling of the bacterial luciferase-flavin mononucleotide complex combining flexible docking with structure-activity data
Abstract
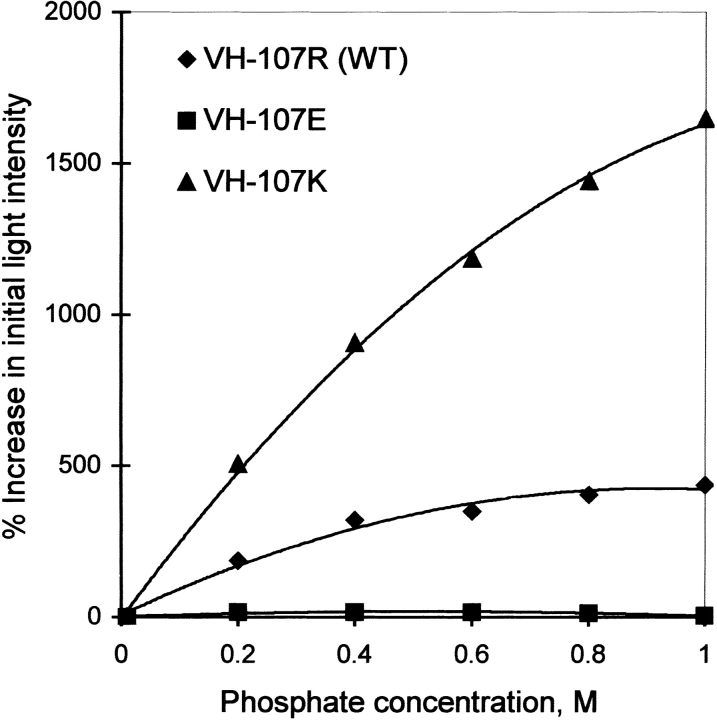
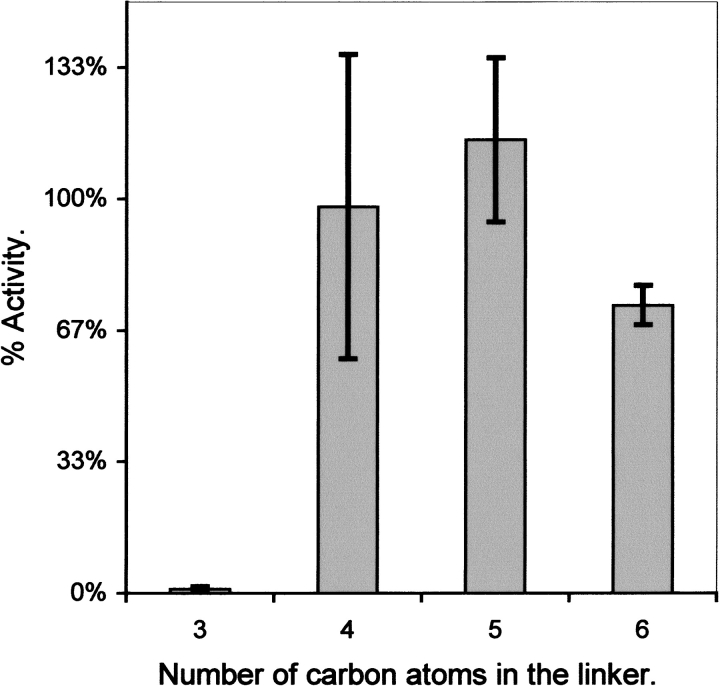
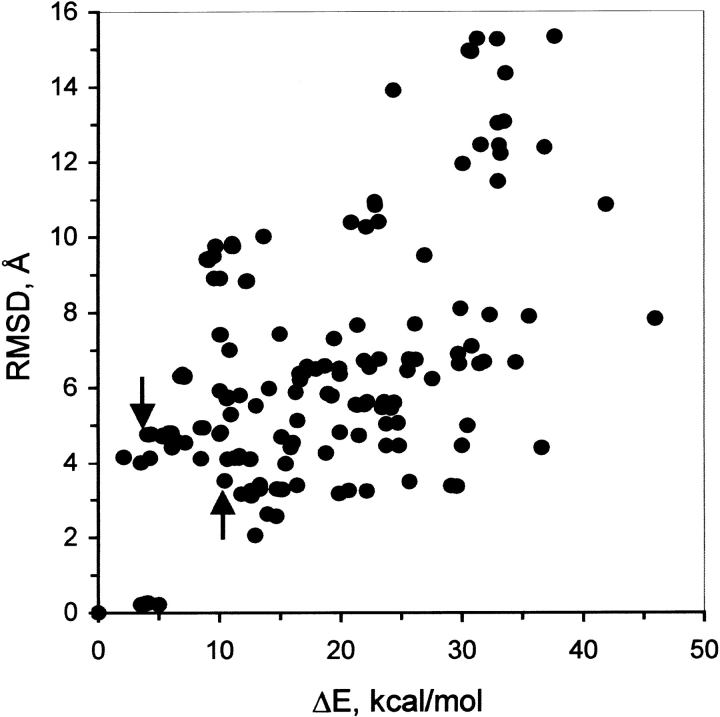
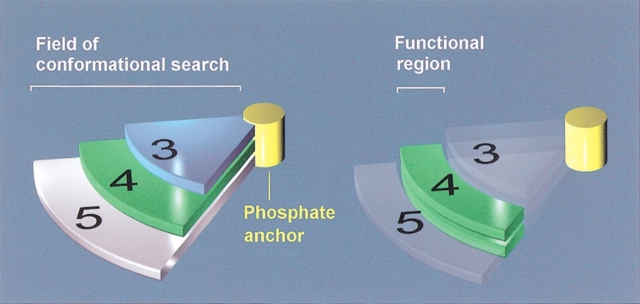
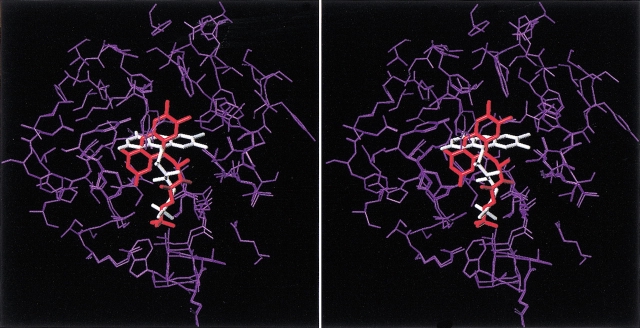
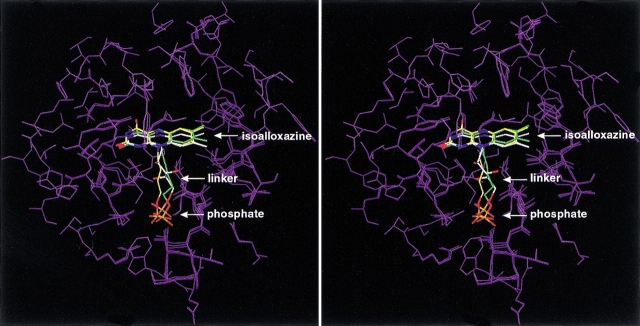
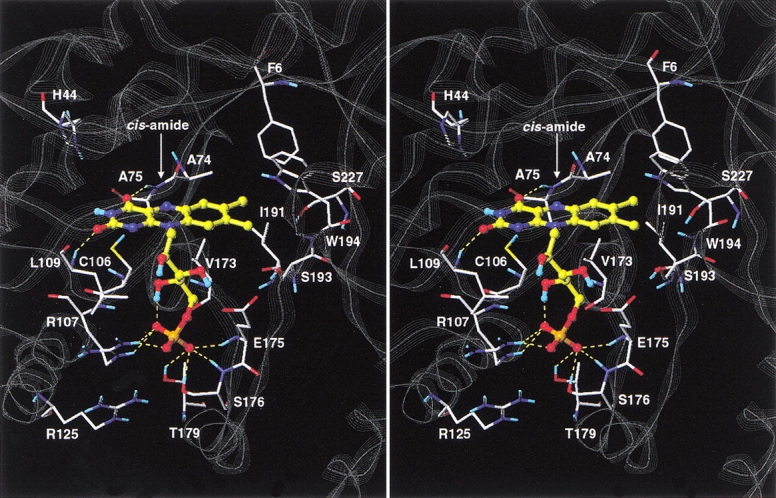
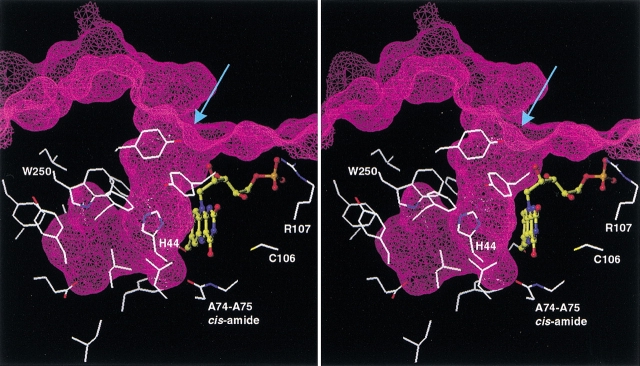
Although the crystal structure of Vibrio harveyi luciferase has been elucidated, the binding sites for the flavin mononucleotide and fatty aldehyde substrates are still unknown. The determined location of the phosphate-binding site close to Arg 107 on the alpha subunit of luciferase is supported here by point mutagenesis. This information, together with previous structure-activity data for the length of the linker connecting the phosphate group to the isoalloxazine ring represent important characteristics of the luciferase-bound conformation of the flavin mononucleotide. A model of the luciferase-flavin complex is developed here using flexible docking supplemented by these structural constraints. The location of the phosphate moiety was used as the anchor in a flexible docking procedure performed by conformation search by using the Monte Carlo minimization approach. The resulting databases of energy-ranked feasible conformations of the luciferase complexes with flavin mononucleotide, omega-phosphopentylflavin, omega-phosphobutylflavin, and omega-phosphopropylflavin were filtered according to the structure-activity profile of these analogs. A unique model was sought not only on energetic criteria but also on the geometric requirement that the isoalloxazine ring of the active flavin analogs must assume a common orientation in the luciferase-binding site, an orientation that is also inaccessible to the inactive flavin analog. The resulting model of the bacterial luciferase-flavin mononucleotide complex is consistent with the experimental data available in the literature. Specifically, the isoalloxazine ring of the flavin mononucleotide interacts with the Ala 74-Ala 75 cis-peptide bond as well as with the Cys 106 side chain in the alpha subunit of luciferase. The model of the binary complex reveals a distinct cavity suitable for aldehyde binding adjacent to the isoalloxazine ring and flanked by other key residues (His 44 and Trp 250) implicated in the active site.
Figures
References
-
- AbouKhair, N.K., Ziegler, M.M., and Baldwin, T.O. 1985. Bacterial luciferase: Demonstration of a catalytically competent altered conformational state following a single turnover. Biochemistry 24 3942–3947. - PubMed
-
- Abu-Soud, H.M., Clark, A.C., Francisco, W.A., Baldwin, T.O., and Raushel, F.M. 1993. Kinetic destabilization of the hydroperoxy flavin intermediate by site-directed modification of the reactive thiol in bacterial luciferase. J. Biol. Chem. 268 7699–7706. - PubMed
-
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, Jr. K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of protein, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117 5179–5197.
-
- Chen, L.H. and Baldwin, T.O. 1989. Random and site-directed mutagenesis of bacterial luciferase: Investigation of the aldehyde binding site. Biochemistry 28 2684–2689. - PubMed
-
- Cline, T.W. and Hastings, J.W. 1972. Mutationally altered bacterial luciferase. Implications for subunit functions. Biochemistry 11 3359–3370. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
