

Evolutionary self-organization of cell-free genetic coding
- PMID: 11470896
- PMCID: PMC55395
- DOI: 10.1073/pnas.151253198
Evolutionary self-organization of cell-free genetic coding
Abstract
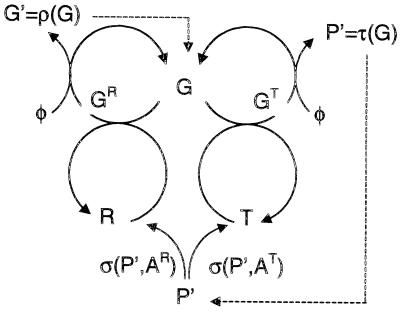
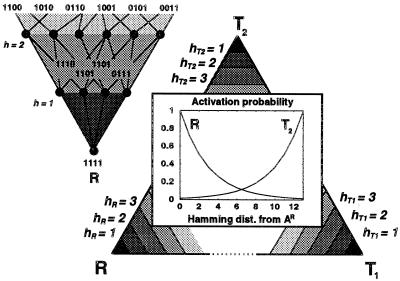
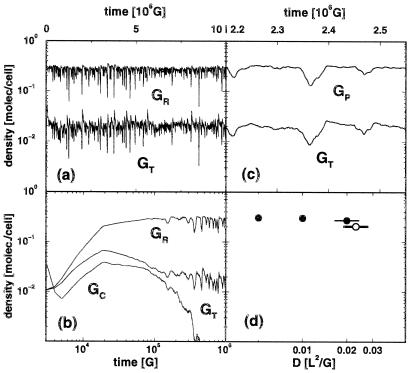
Genetic encoding provides a generic construction scheme for biomolecular functions. This paper addresses the key problem of coevolution and exploitation of the multiple components necessary to implement a replicable genetic encoding scheme. Extending earlier results on multicomponent replication, the necessity of spatial structure for the evolutionary stabilization of the genetic coding system is established. An individual-based stochastic model of interacting molecules in three-dimensional space is presented that allows the evolution of genetic coding to be analyzed explicitly. A massively parallel configurable computer (NGEN) is used to implement the model, on the time scale of millions of generations, directly in electronic hardware. The spatial correlations between components of the genetic coding system are analyzed and found to be essential for evolutionary stability.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
