

16S/23S rRNA intergenic spacer regions for phylogenetic analysis, identification, and subtyping of Bartonella species
- PMID: 11473990
- PMCID: PMC88237
- DOI: 10.1128/JCM.39.8.2768-2778.2001
16S/23S rRNA intergenic spacer regions for phylogenetic analysis, identification, and subtyping of Bartonella species
Abstract
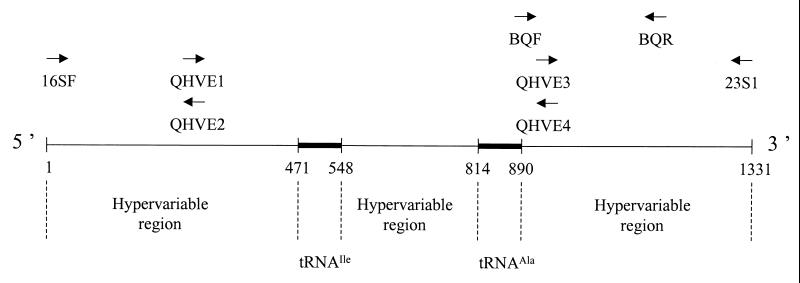
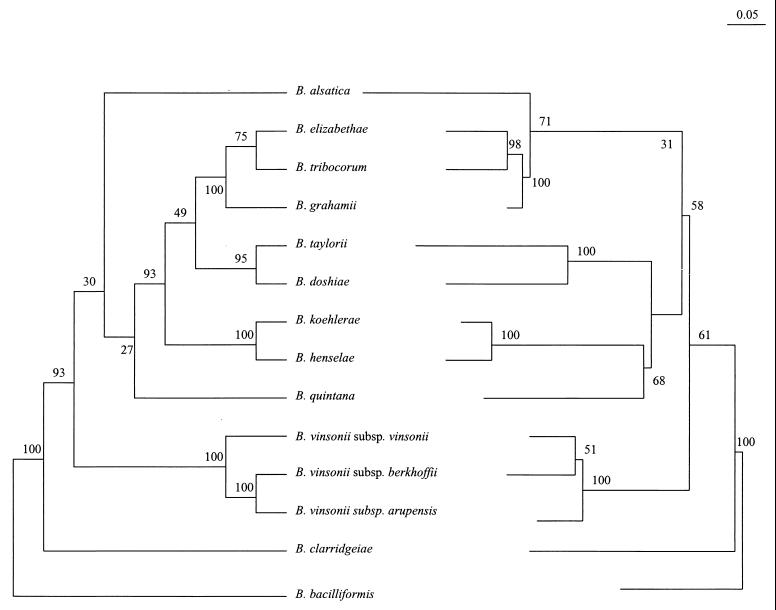
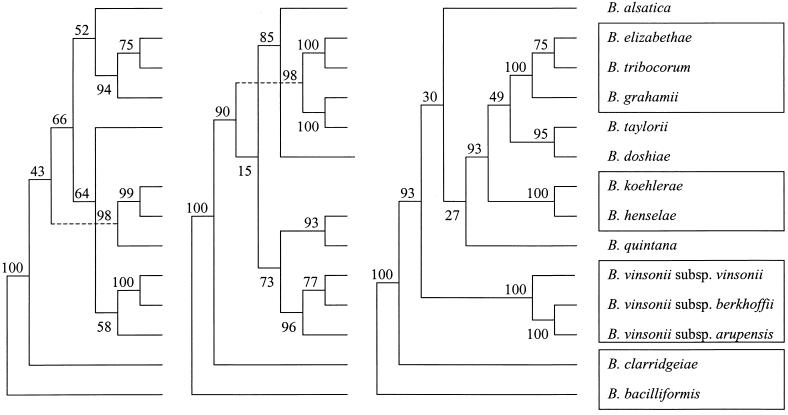
Species of the genus Bartonella are currently recognized in growing numbers and are involved in an increasing variety of human diseases, mainly trench fever, Carrion's disease, bacillary angiomatosis, endocarditis, cat scratch disease, neuroretinitis, and asymptomatic bacteremia. Such a wide spectrum of infections makes it necessary to develop species and strain identification tools in order to perform phylogenetic and epidemiological studies. The 16S/23S rRNA intergenic spacer region (ITS) was sequenced for four previously untested species, B. vinsonii subsp. arupensis, B. tribocorum, B. alsatica, and B. koehlerae, as well as for 28 human isolates of B. quintana (most of them from French homeless people), six human or cat isolates of B. henselae, five cat isolates of B. clarridgeiae, and four human isolates of B. bacilliformis. Phylogenetic trees inferred from full ITS sequences of the 14 recognized Bartonella species using parsimony and distance methods revealed high statistical support, as bootstrap values were higher than those observed with other tested genes. Five well-supported lineages were identified within the genus and the proposed phylogenetic organization was consistent with that resulting from protein-encoding gene sequence comparisons. The ITS-derived phylogeny appears, therefore, to be a useful tool for investigating the evolutionary relationships of Bartonella species and to identify Bartonella species. Further, partial ITS amplification and sequencing offers a sensitive means of intraspecies differentiation of B. henselae, B. clarridgeiae, and B. bacilliformis isolates, as each strain had a specific sequence. The usefulness of this approach in epidemiological investigations should be highlighted. Among B. quintana strains, however, the genetic heterogeneity was low, as only three ITS genotypes were identified. It was nevertheless sufficient to show that the B. quintana population infecting homeless people in France was not clonal.
Figures


Similar articles
-
Inter- and intraspecies identification of Bartonella (Rochalimaea) species.J Clin Microbiol. 1995 Jun;33(6):1573-9. doi: 10.1128/jcm.33.6.1573-1579.1995. J Clin Microbiol. 1995. PMID: 7650189 Free PMC article.
-
Genetic classification and differentiation of Bartonella species based on comparison of partial ftsZ gene sequences.J Clin Microbiol. 2002 Oct;40(10):3641-7. doi: 10.1128/JCM.40.10.3641-3647.2002. J Clin Microbiol. 2002. PMID: 12354859 Free PMC article.
-
Phylogenetic classification of Bartonella species by comparing groEL sequences.Int J Syst Evol Microbiol. 2002 Jan;52(Pt 1):165-71. doi: 10.1099/00207713-52-1-165. Int J Syst Evol Microbiol. 2002. PMID: 11837299
-
Molecular phylogeny of the genus Bartonella: what is the current knowledge?FEMS Microbiol Lett. 2001 Jun 12;200(1):1-7. doi: 10.1111/j.1574-6968.2001.tb10684.x. FEMS Microbiol Lett. 2001. PMID: 11410341 Review.
-
[Bartonellosis].Recenti Prog Med. 2003 Apr;94(4):177-85. Recenti Prog Med. 2003. PMID: 12677790 Review. Italian.
Cited by
-
Blood culture-negative endocarditis caused by Bartonella quintana in Iran.Sci Rep. 2024 Oct 30;14(1):26063. doi: 10.1038/s41598-024-77757-0. Sci Rep. 2024. PMID: 39478136 Free PMC article.
-
Bartonella quintana infections in captive monkeys, China.Emerg Infect Dis. 2011 Sep;17(9):1707-9. doi: 10.3201/eid1709.110133. Emerg Infect Dis. 2011. PMID: 21888799 Free PMC article.
-
First Case of Bartonella henselae Bacteremia in Korea.Infect Chemother. 2013 Dec;45(4):446-50. doi: 10.3947/ic.2013.45.4.446. Epub 2013 Dec 27. Infect Chemother. 2013. PMID: 24475360 Free PMC article.
-
Bartonella vinsonii subsp. arupensis infection in animals of veterinary importance, ticks and biopsy samples.New Microbes New Infect. 2020 Jan 16;34:100652. doi: 10.1016/j.nmni.2020.100652. eCollection 2020 Mar. New Microbes New Infect. 2020. PMID: 32071727 Free PMC article.
-
Recombination drives evolution of the Clostridium difficile 16S-23S rRNA intergenic spacer region.PLoS One. 2014 Sep 15;9(9):e106545. doi: 10.1371/journal.pone.0106545. eCollection 2014. PLoS One. 2014. PMID: 25222120 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
