

A missense mutation (Q279R) in the fumarylacetoacetate hydrolase gene, responsible for hereditary tyrosinemia, acts as a splicing mutation
- PMID: 11476670
- PMCID: PMC35353
- DOI: 10.1186/1471-2156-2-9
A missense mutation (Q279R) in the fumarylacetoacetate hydrolase gene, responsible for hereditary tyrosinemia, acts as a splicing mutation
Abstract
Background: Tyrosinemia type I, the most severe disease of the tyrosine catabolic pathway is caused by a deficiency in fumarylacetoacetate hydrolase (FAH). A patient showing few of the symptoms associated with the disease, was found to be a compound heterozygote for a splice mutation, IVS6-1g->t, and a putative missense mutation, Q279R. Analysis of FAH expression in liver sections obtained after resection for hepatocellular carcinoma revealed a mosaic pattern of expression. No FAH was found in tumor regions while a healthy region contained enzyme-expressing nodules.
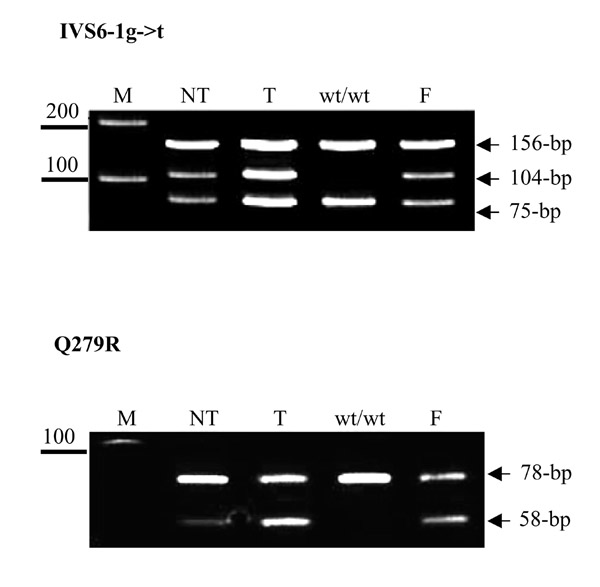
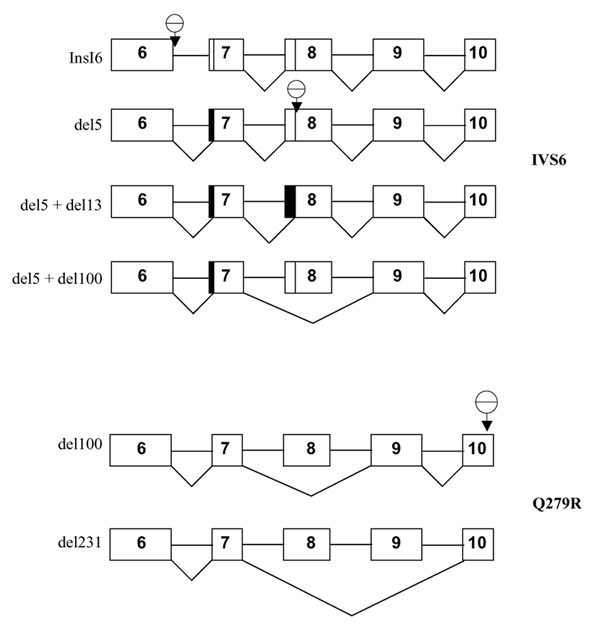
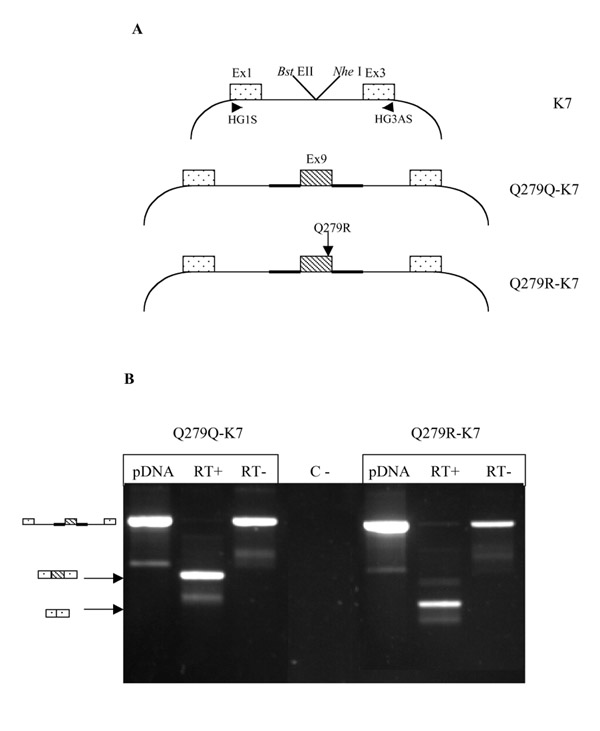
Results: Analysis of DNA from a FAH expressing region showed that the expression of the protein was due to correction of the Q279R mutation. RT-PCR was used to assess if Q279R RNA was produced in the liver cells and in fibroblasts from the patient. Normal mRNA was found in the liver region where the mutation had reverted while splicing intermediates were found in non-expressing regions suggesting that the Q279R mutation acted as a splicing mutation in vivo. Sequence of transcripts showed skipping of exon 8 alone or together with exon 9. Using minigenes in transfection assays, the Q279R mutation was shown to induce skipping of exon 9 when placed in a constitutive splicing environment.
Conclusion: These data suggest that the putative missense mutation Q279R in the FAH gene acts as a splicing mutation in vivo. Moreover FAH expression can be partially restored in certain liver cells as a result of a reversion of the Q279R mutation and expansion of the corrected cells.
Figures

Similar articles
-
Presence of three mutations in the fumarylacetoacetate hydrolase gene in a patient with atypical symptoms of hereditary tyrosinemia type I.Mol Genet Metab. 2019 May;127(1):58-63. doi: 10.1016/j.ymgme.2019.01.019. Epub 2019 Feb 7. Mol Genet Metab. 2019. PMID: 30954369
-
Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1.Proc Natl Acad Sci U S A. 2001 Jan 16;98(2):641-5. doi: 10.1073/pnas.98.2.641. Proc Natl Acad Sci U S A. 2001. PMID: 11209059 Free PMC article.
-
Structural and functional analysis of missense mutations in fumarylacetoacetate hydrolase, the gene deficient in hereditary tyrosinemia type 1.J Biol Chem. 2001 May 4;276(18):15225-31. doi: 10.1074/jbc.M009341200. Epub 2001 Jan 22. J Biol Chem. 2001. PMID: 11278491
-
Molecular Aspects of the FAH Mutations Involved in HT1 Disease.Adv Exp Med Biol. 2017;959:25-48. doi: 10.1007/978-3-319-55780-9_3. Adv Exp Med Biol. 2017. PMID: 28755182 Review.
-
Biochemical and Clinical Aspects of Hereditary Tyrosinemia Type 1.Adv Exp Med Biol. 2017;959:9-21. doi: 10.1007/978-3-319-55780-9_2. Adv Exp Med Biol. 2017. PMID: 28755181 Review.
Cited by
-
Geographical and Ethnic Distribution of Mutations of the Fumarylacetoacetate Hydrolase Gene in Hereditary Tyrosinemia Type 1.JIMD Rep. 2015;19:43-58. doi: 10.1007/8904_2014_363. Epub 2015 Feb 15. JIMD Rep. 2015. PMID: 25681080 Free PMC article.
-
In vivo reversion to normal of inherited mutations in humans.J Med Genet. 2003 Oct;40(10):721-8. doi: 10.1136/jmg.40.10.721. J Med Genet. 2003. PMID: 14569115 Free PMC article. Review.
-
Somatic genetic rescue in Mendelian haematopoietic diseases.Nat Rev Genet. 2019 Oct;20(10):582-598. doi: 10.1038/s41576-019-0139-x. Epub 2019 Jun 11. Nat Rev Genet. 2019. PMID: 31186537 Review.
-
Tyrosinaemia type I--de novo mutation in liver tissue suppressing an inborn splicing defect.J Mol Med (Berl). 2005 May;83(5):406-10. doi: 10.1007/s00109-005-0648-2. Epub 2005 Mar 10. J Mol Med (Berl). 2005. PMID: 15759101
-
A minor alternative transcript of the fumarylacetoacetate hydrolase gene produces a protein despite being likely subjected to nonsense-mediated mRNA decay.BMC Mol Biol. 2005 Jan 7;6:1. doi: 10.1186/1471-2199-6-1. BMC Mol Biol. 2005. PMID: 15638932 Free PMC article.
References
-
- Tanguay RM, Laberge C, Lescault A, Valet JP, Duband JL, Quenneville Y. Molecular basis of hereditary tyrosinemias: Proof of the primary defect by western blot. Advances in Gene technology: Human genetic disorders (Edited by Ahmad F, Black S, Schultz J, Scott WA, Whelan WJ) Cambridge : University Press, Cambridge. 1984:256–257.
-
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. Metabolic and Molecular Bases of Inherited Disease, 8th Ed. (Edited by Scriver CR, Beaudet AL, Sly WS, Valle D) McGraw-Hill, Baltimore. 2001:1777–1805.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
