

Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system
- PMID: 11483577
- PMCID: PMC311082
- DOI: 10.1101/gr.181001
Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system
Abstract
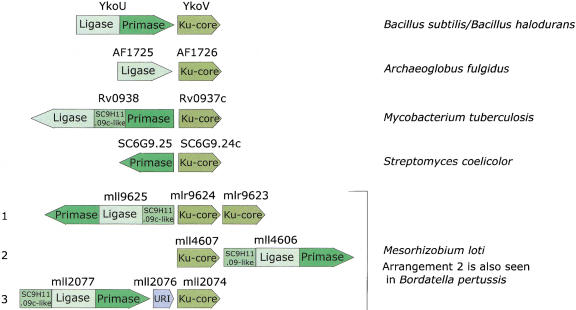
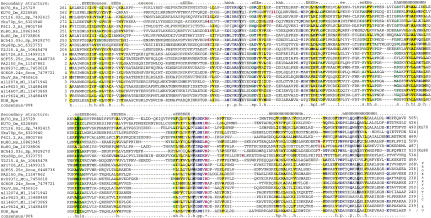
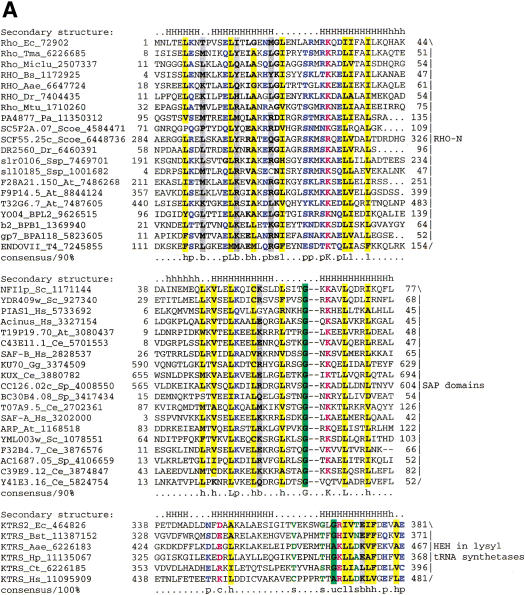
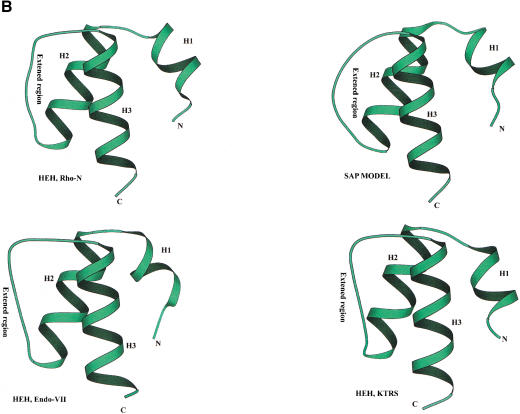
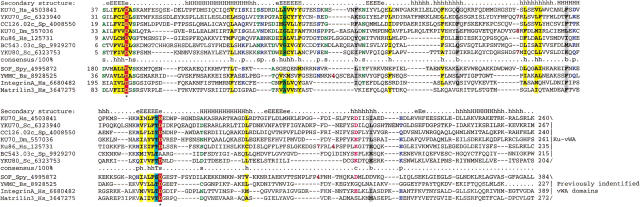
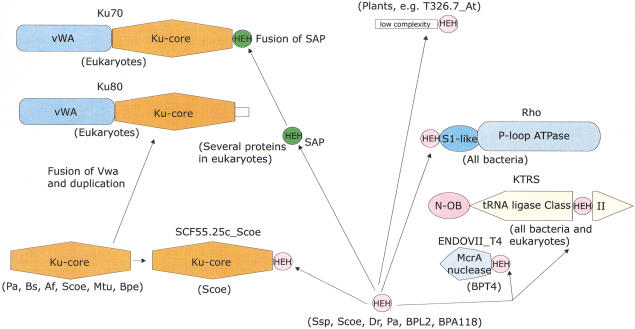
Homologs of the eukaryotic DNA-end-binding protein Ku were identified in several bacterial and one archeal genome using iterative database searches with sequence profiles. Identification of prokaryotic Ku homologs allowed the dissection of the Ku protein sequences into three distinct domains, the Ku core that is conserved in eukaryotes and prokaryotes, a derived von Willebrand A domain that is fused to the amino terminus of the core in eukaryotic Ku proteins, and the newly recognized helix-extension-helix (HEH) domain that is fused to the carboxyl terminus of the core in eukaryotes and in one of the Ku homologs from the Actinomycete Streptomyces coelicolor. The version of the HEH domain present in eukaryotic Ku proteins represents the previously described DNA-binding domain called SAP. The Ku homolog from S. coelicolor contains a distinct version of the HEH domain that belongs to a previously unnoticed family of nucleic-acid-binding domains, which also includes HEH domains from the bacterial transcription termination factor Rho, bacterial and eukaryotic lysyl-tRNA synthetases, bacteriophage T4 endonuclease VII, and several uncharacterized proteins. The distribution of the Ku homologs in bacteria coincides with that of the archeal-eukaryotic-type DNA primase and genes for prokaryotic Ku homologs form predicted operons with genes coding for an ATP-dependent DNA ligase and/or archeal-eukaryotic-type DNA primase. Some of these operons additionally encode an uncharacterized protein that may function as nuclease or an Slx1p-like predicted nuclease containing a URI domain. A hypothesis is proposed that the Ku homolog, together with the associated gene products, comprise a previously unrecognized prokaryotic system for repair of double-strand breaks in DNA.
Figures


References
-
- Allison TJ, Wood TC, Briercheck DM, Rastinejad F, Richardson JP, Rule GS. Crystal structure of the RNA-binding domain from transcription termination factor rho [letter] Nat Struct Biol. 1998;5:352–356. - PubMed
-
- ————— SAP — a putative DNA-binding motif involved in chromosomal organization. Trends Biochem Sci. 2000;25:112–114. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous