

Salicylates inhibit flavivirus replication independently of blocking nuclear factor kappa B activation
- PMID: 11483726
- PMCID: PMC115025
- DOI: 10.1128/jvi.75.17.7828-7839.2001
Salicylates inhibit flavivirus replication independently of blocking nuclear factor kappa B activation
Abstract
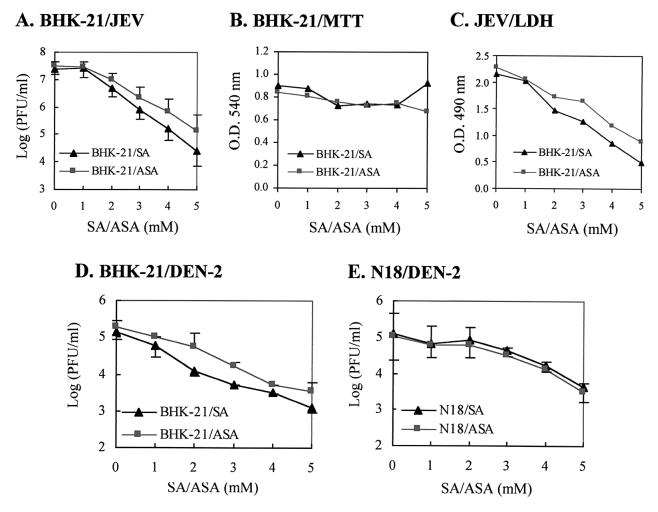
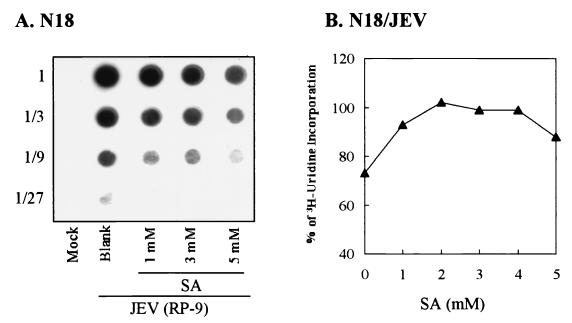
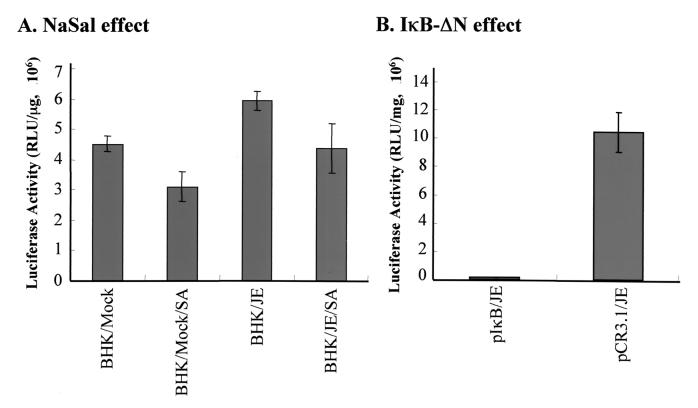
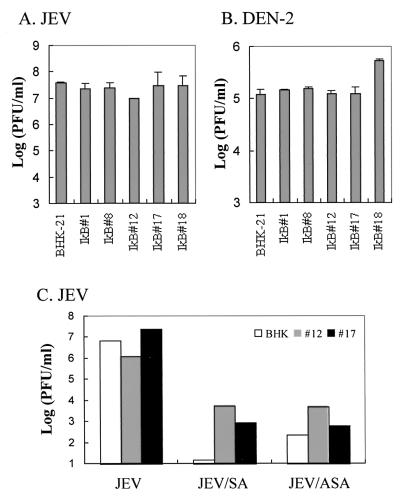
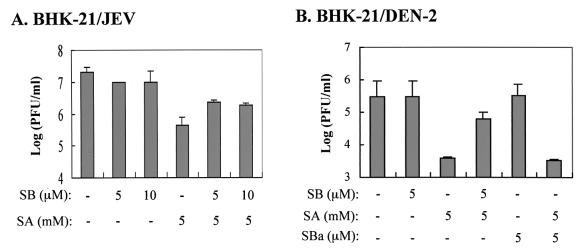
Flaviviruses comprise a positive-sense RNA genome that replicates exclusively in the cytoplasm of infected cells. Whether flaviviruses require an activated nuclear factor(s) to complete their life cycle and trigger apoptosis in infected cells remains elusive. Flavivirus infections quickly activate nuclear factor kappa B (NF-kappaB), and salicylates have been shown to inhibit NF-kappaB activation. In this study, we investigated whether salicylates suppress flavivirus replication and virus-induced apoptosis in cultured cells. In a dose-dependent inhibition, we found salicylates within a range of 1 to 5 mM not only restricted flavivirus replication but also abrogated flavivirus-triggered apoptosis. However, flavivirus replication was not affected by a specific NF-kappaB peptide inhibitor, SN50, and a proteosome inhibitor, lactacystin. Flaviviruses also replicated and triggered apoptosis in cells stably expressing IkappaBalpha-DeltaN, a dominant-negative mutant that antagonizes NF-kappaB activation, as readily as in wild-type BHK-21 cells, suggesting that NF-kappaB activation is not essential for either flavivirus replication or flavivirus-induced apoptosis. Salicylates still diminished flavivirus replication and blocked apoptosis in the same IkappaBalpha-DeltaN cells. This inhibition of flaviviruses by salicylates could be partially reversed by a specific p38 mitogen-activated protein (MAP) kinase inhibitor, SB203580. Together, these results show that the mechanism by which salicylates suppress flavivirus infection may involve p38 MAP kinase activity but is independent of blocking the NF-kappaB pathway.
Figures



References
-
- Avirutnan P, Malasit P, Seliger B, Bhakdi S, Husmann M. Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J Immunol. 1998;161:6338–6346. - PubMed
-
- Baeuerle P A, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20.20. - PubMed
-
- Baeuerle P A, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. - PubMed
-
- Barkett M, Gilmore T D. Control of apoptosis by Rel/NF-κB transcription factors. Oncogene. 1999;18:6910–6924. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
