

Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens
- PMID: 11489948
- PMCID: PMC2193475
- DOI: 10.1084/jem.194.3.285
Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens
Abstract
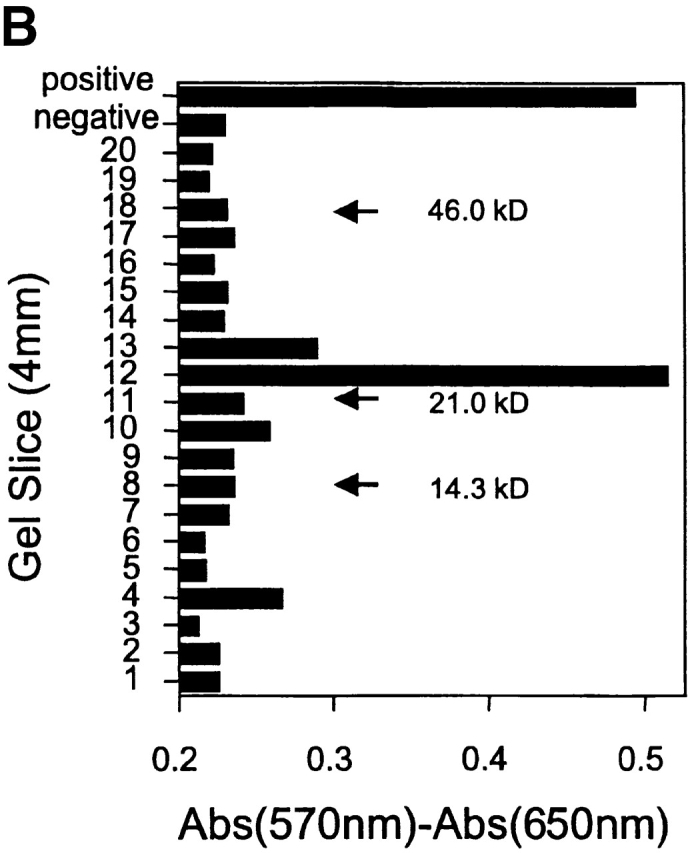
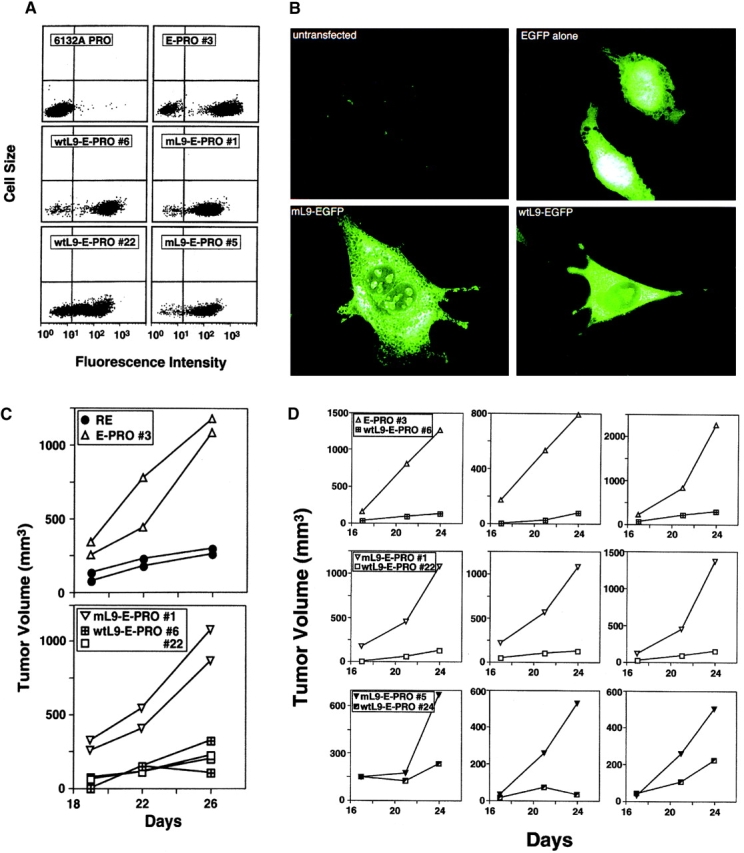
Antigens that are tumor specific yet retained by tumor cells despite tumor progression offer stable and specific targets for immunologic and possibly other therapeutic interventions. Therefore, we have studied two CD4(+) T cell-recognized tumor-specific antigens that were retained during evolution of two ultraviolet-light-induced murine cancers to more aggressive growth. The antigens are ribosomal proteins altered by somatic tumor-specific point mutations, and the progressor (PRO) variants lack the corresponding normal alleles. In the first tumor, 6132A-PRO, the antigen is encoded by a point-mutated L9 ribosomal protein gene. The tumor lacks the normal L9 allele because of an interstitial deletion from chromosome 5. In the second tumor, 6139B-PRO, both alleles of the L26 gene have point mutations, and each encodes a different tumor-specific CD4(+) T cell-recognized antigen. Thus, for both L9 and L26 genes, we observe "two hit" kinetics commonly observed in genes suppressing tumor growth. Indeed, reintroduction of the lost wild-type L9 allele into the 6132A-PRO variant suppressed the growth of the tumor cells in vivo. Since both L9 and L26 encode proteins essential for ribosomal biogenesis, complete loss of the tumor-specific target antigens in the absence of a normal allele would abrogate tumor growth.
Figures










References
-
- Monach P.A., Meredith S.C., Siegel C.T., Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. - PubMed
-
- Wölfel T., Hauer M., Schneider J., Serrano M., Wölfel C., Klehmann-Hieb E., De Plaen E., Hankeln T., Meyer zum Buschenfelde K.H., Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
