

Optimal conditions to use Pfu exo(-) DNA polymerase for highly efficient ligation-mediated polymerase chain reaction protocols
- PMID: 11504891
- PMCID: PMC55867
- DOI: 10.1093/nar/29.16.e83
Optimal conditions to use Pfu exo(-) DNA polymerase for highly efficient ligation-mediated polymerase chain reaction protocols
Abstract
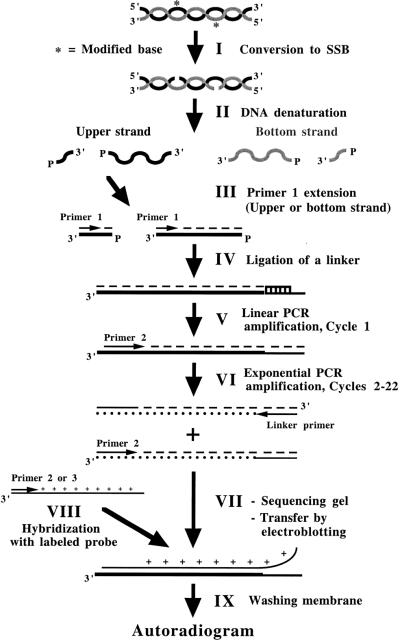
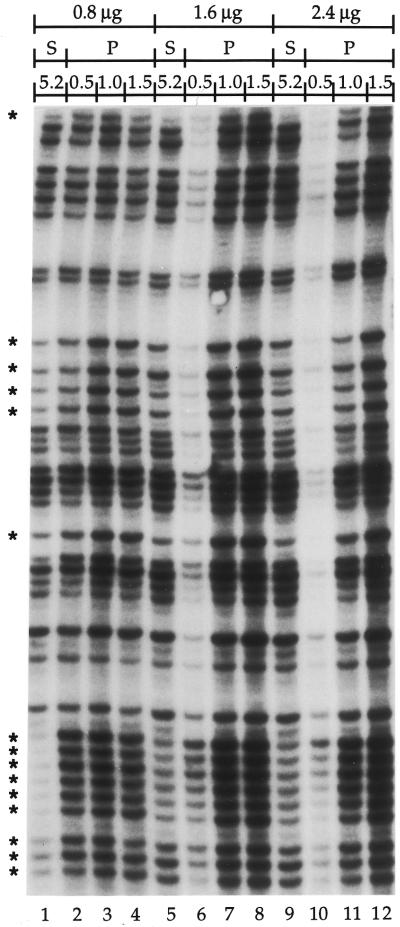
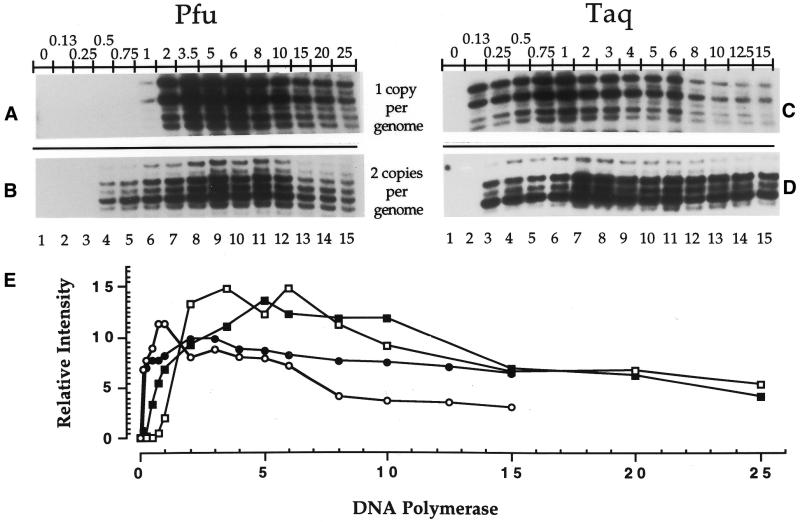
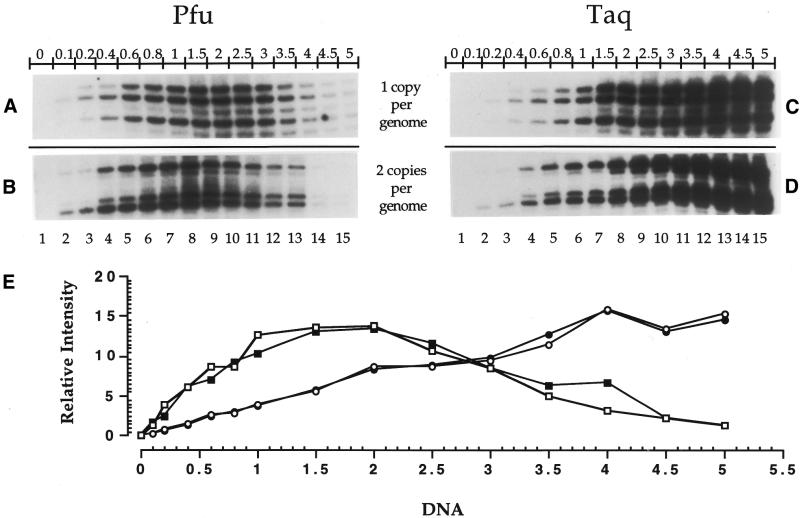
Ligation-Mediated Polymerase Chain Reaction (LMPCR) is the most sensitive sequencing technique available to map single-stranded DNA breaks at the nucleotide level of resolution using genomic DNA. LMPCR has been adapted to map DNA damage and reveal DNA-protein interactions inside living cells. However, the sequence context (GC content), the global break frequency and the current combination of DNA polymerases used in LMPCR affect the quality of the results. In this study, we developed and optimized an LMPCR protocol adapted for Pyrococcus furiosus exo(-) DNA polymerase (Pfu exo(-)). The relative efficiency of Pfu exo(-) was compared to T7-modified DNA polymerase (Sequenase 2.0) at the primer extension step and to Thermus aquaticus DNA polymerase (Taq) at the PCR amplification step of LMPCR. At all break frequencies tested, Pfu exo(-) proved to be more efficient than Sequenase 2.0. During both primer extension and PCR amplification steps, the ratio of DNA molecules per unit of DNA polymerase was the main determinant of the efficiency of Pfu exo(-), while the efficiency of Taq was less affected by this ratio. Substitution of NaCl for KCl in the PCR reaction buffer of Taq strikingly improved the efficiency of the DNA polymerase. Pfu exo(-) was clearly more efficient than Taq to specifically amplify extremely GC-rich genomic DNA sequences. Our results show that a combination of Pfu exo(-) at the primer extension step and Taq at the PCR amplification step is ideal for in vivo DNA analysis and DNA damage mapping using LMPCR.
Figures
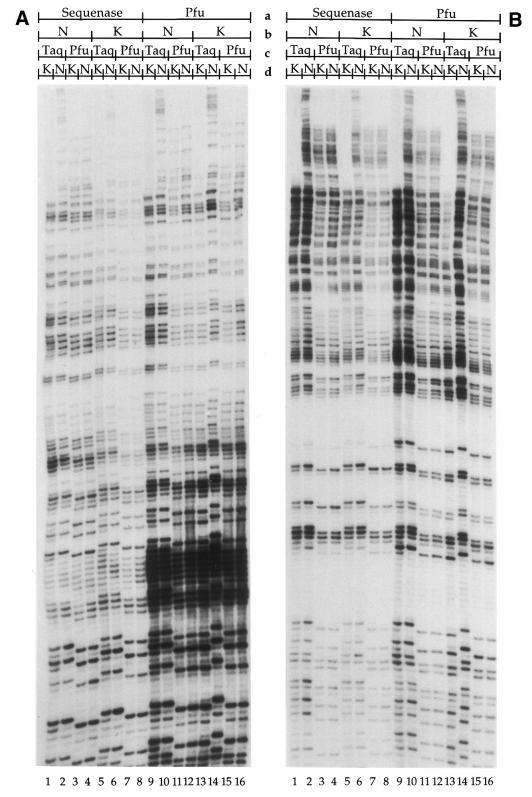
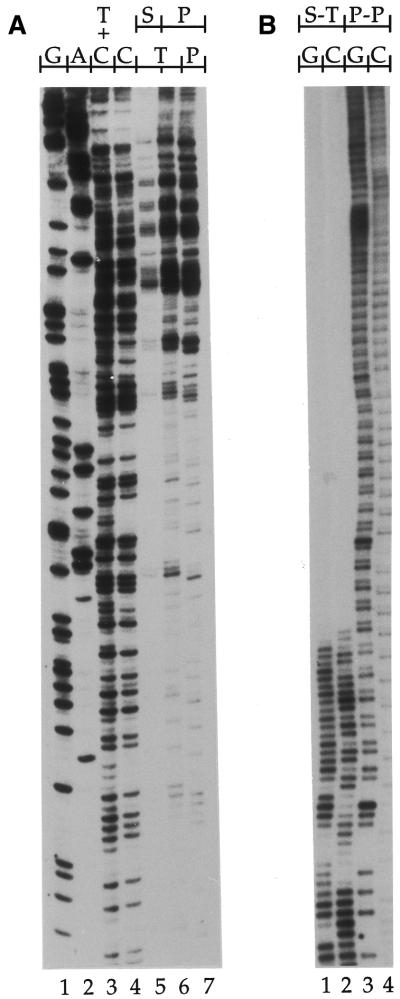
References
-
- Pfeifer G.P., Singer-Sam,J. and Riggs,A.D. (1993) Analysis of methylation and chromatin structure. Methods Enzymol., 225, 567–583. - PubMed
-
- Cartwright I.L. and Kelly,S.E. (1991) Probing the nature of chromosomal DNA–protein contacts by in vivo footprinting. Biotechniques, 11, 188–196. - PubMed
-
- Mueller P.R. and Wold,B. (1989) In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science, 246, 780–786. [Published erratum appears in Science (1990) 248, 802] - PubMed
-
- Pfeifer G.P., Steigerwald,S.D., Mueller,P.R., Wold,B. and Riggs,A.D. (1989) Genomic sequencing and methylation analysis by ligation mediated PCR. Science, 246, 810–813. - PubMed
-
- Pfeifer G.P., Tanguay,R.L., Steigerwald,S.D. and Riggs,A.D. (1990) In vivo footprint and methylation analysis by PCR-aided genomic sequencing: comparison of active and inactive X chromosomal DNA at the CpG island and promoter of human PGK-1. Genes Dev., 4, 1277–1287. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
