

Maple syrup urine disease: identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population
- PMID: 11509994
- PMCID: PMC1226071
- DOI: 10.1086/323677
Maple syrup urine disease: identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population
Abstract
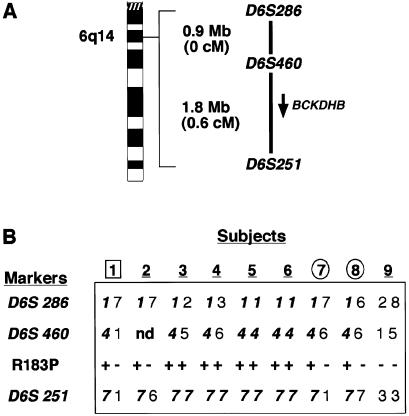
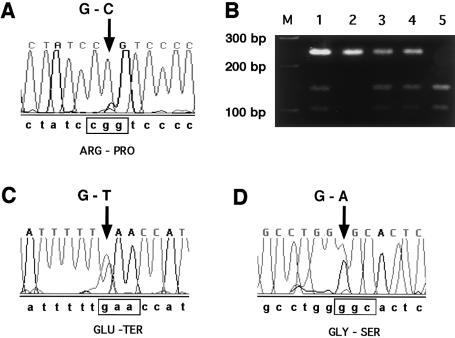
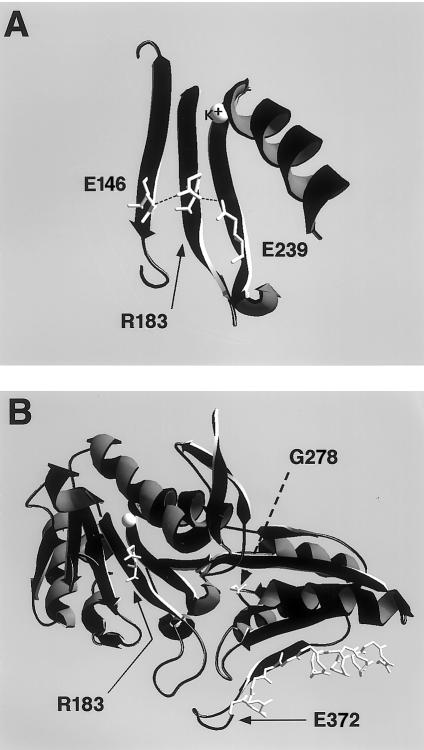
Maple syrup urine disease (MSUD) is a rare, autosomal recessive disorder of branched-chain amino acid metabolism. We noted that a large proportion (10 of 34) of families with MSUD that were followed in our clinic were of Ashkenazi Jewish (AJ) descent, leading us to search for a common mutation within this group. On the basis of genotyping data suggestive of a conserved haplotype at tightly linked markers on chromosome 6q14, the BCKDHB gene encoding the E1beta subunit was sequenced. Three novel mutations were identified in seven unrelated AJ patients with MSUD. The locations of the affected residues in the crystal structure of the E1beta subunit suggested possible mechanisms for the deleterious effects of these mutations. Large-scale population screening of AJ individuals for R183P, the mutation present in six of seven patients, revealed that the carrier frequency of the mutant allele was approximately 1/113; the patient not carrying R183P had a previously described homozygous mutation in the gene encoding the E2 subunit. These findings suggested that a limited number of mutations might underlie MSUD in the AJ population, potentially facilitating prenatal diagnosis and carrier detection of MSUD in this group.
Figures
References
Electronic-Database Information
-
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for human branched-chain α-keto dehydrogenase E1β-subunit mRNA [accession U50708])
-
- GeneMap'99, http://www.ncbi.nlm.nih.gov/genemap99/
-
- Human Genome Project Working Draft at UCSC, http://genome.ucsc.edu
-
- NCBI Structure (Molecular Modeling Database), http://www.ncbi.nlm.nih.gov/Structure/MMDB/mmdb.shtml (for human BCKAD [accession number 1DTW])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for MSUD types Ia [MIM 248600], Ib [MIM 248611]), and II [MIM 248610])
References
-
- Ævarsson A, Chuang JL, Wynn RM, Turley S, Chuang DT, Hol WG (2000) Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure Fold Des 8:277–291 - PubMed
-
- Auerbach AD (1997) Fanconi anemia: genetic testing in Ashkenazi Jews. Genet Test 1:27–33 - PubMed
-
- Bargal R, Avidan N, Olender T, Ben Asher E, Zeigler M, Raas-Rothschild A, Frumkin A, Ben-Yoseph O, Friedlender Y, Lancet D, Bach G (2001) Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum Mutat 17:397–402 - PubMed
-
- Chuang DT, Shih VE (2000) Disorders of branched chain amino acid and keto acid metabolism. In: Scriver CR, Beaudet A, Sly WL, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 1971–2006
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
