

Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity
- PMID: 11517237
- PMCID: PMC6763071
- DOI: 10.1523/JNEUROSCI.21-17-06480.2001
Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity
Abstract
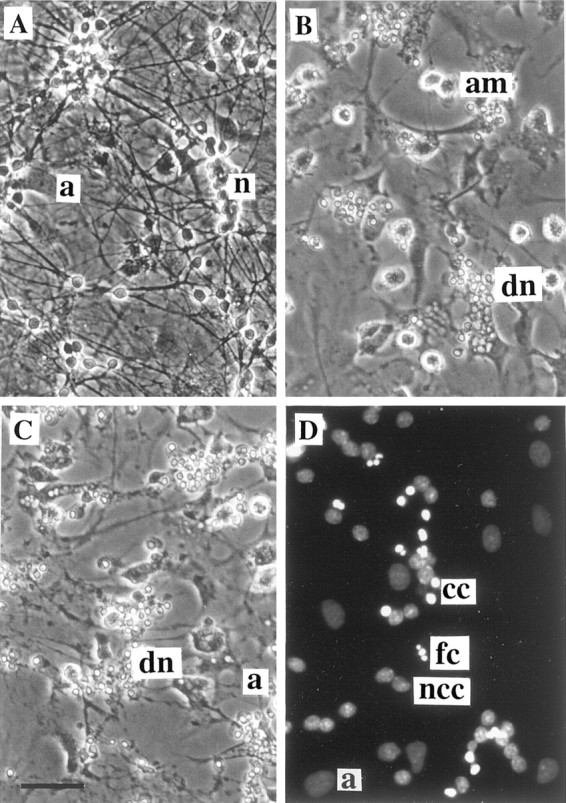
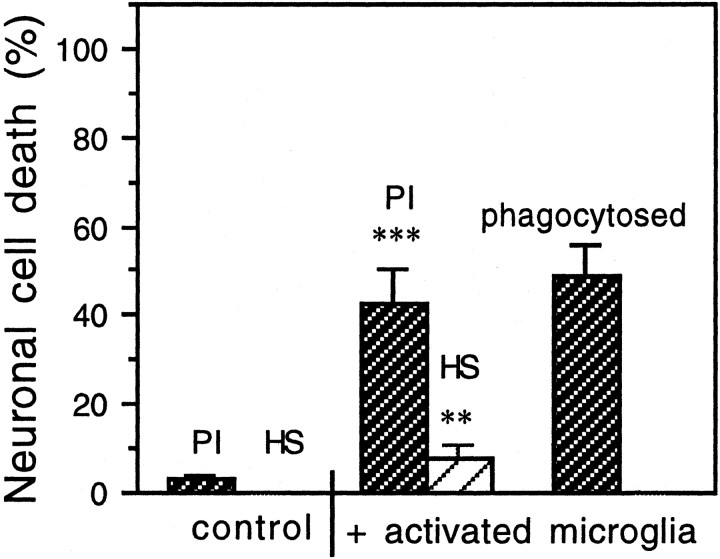
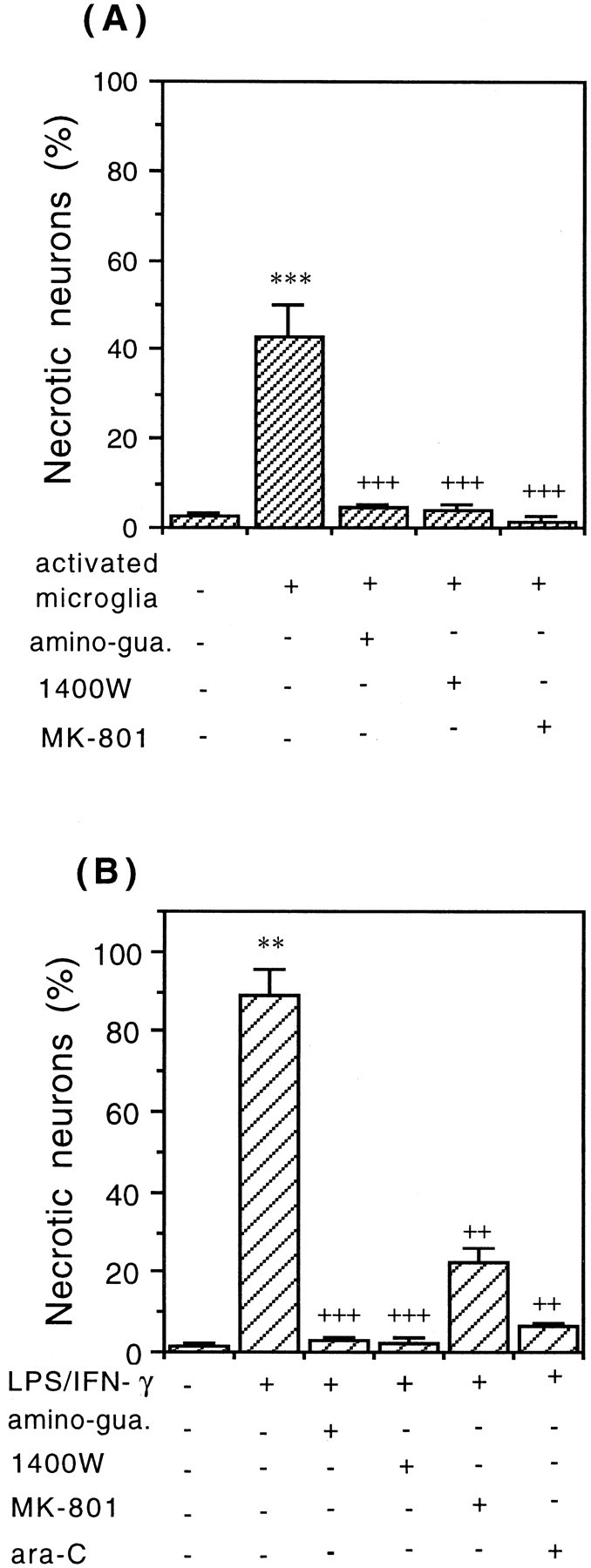
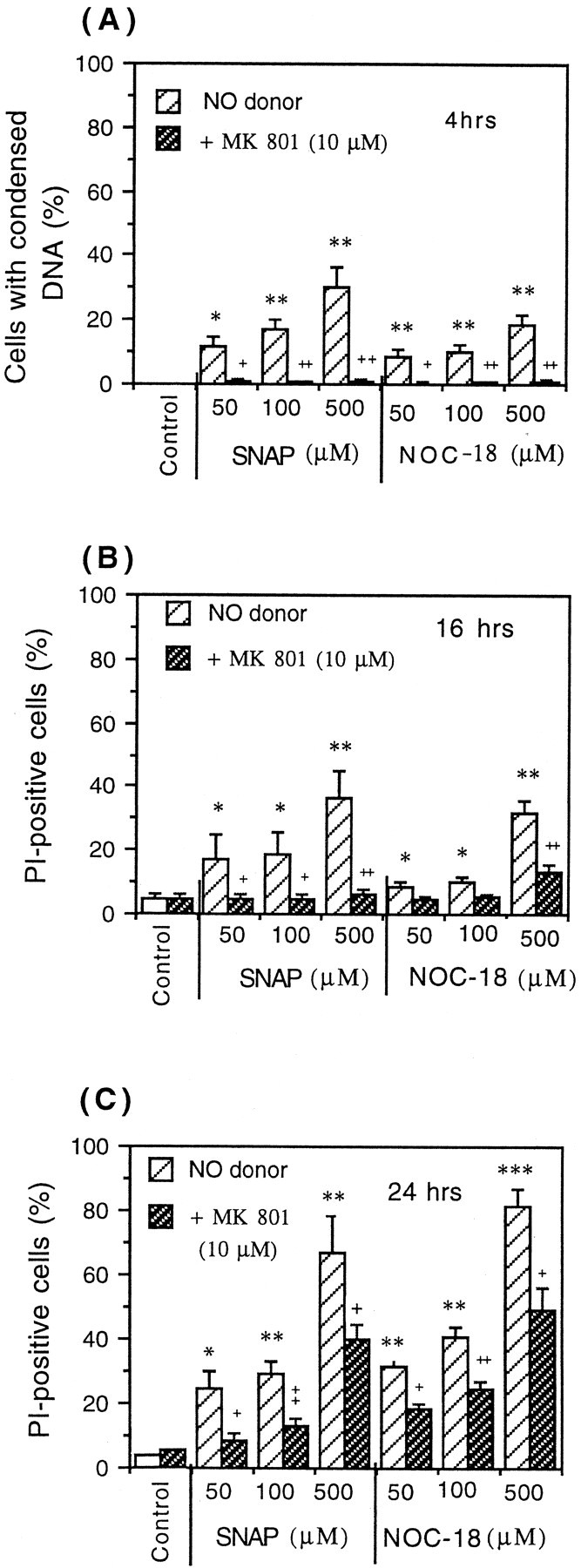
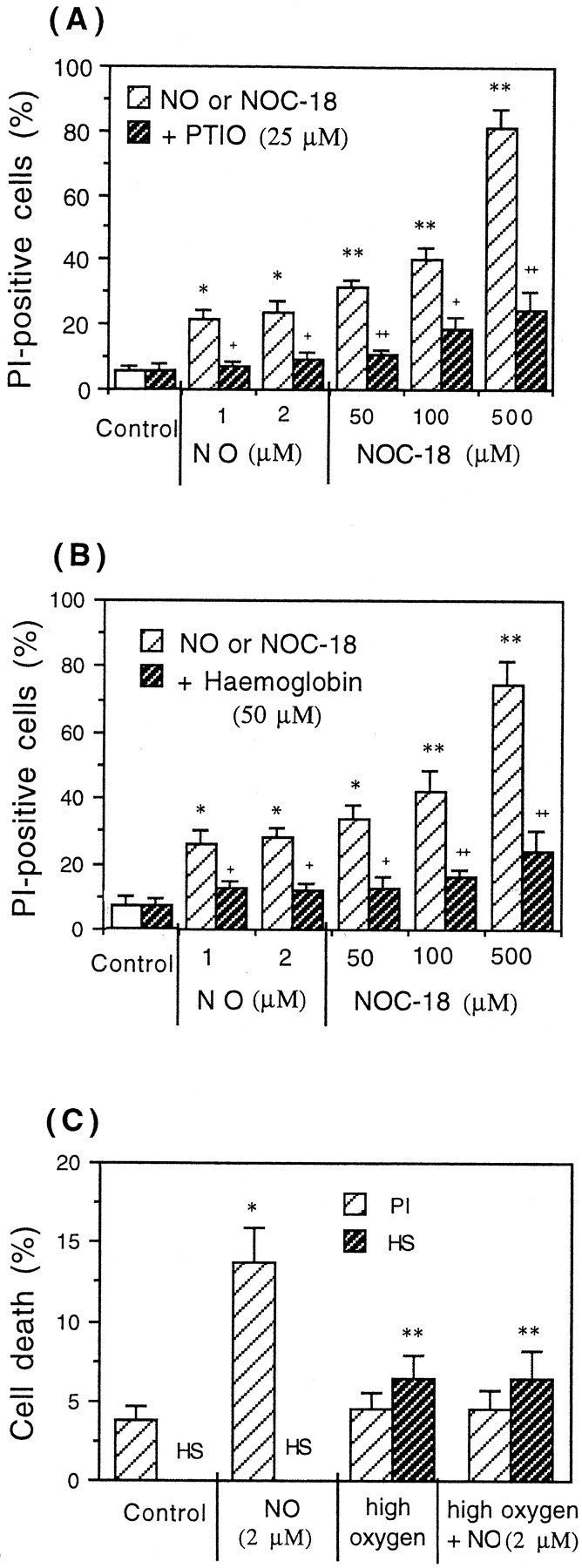
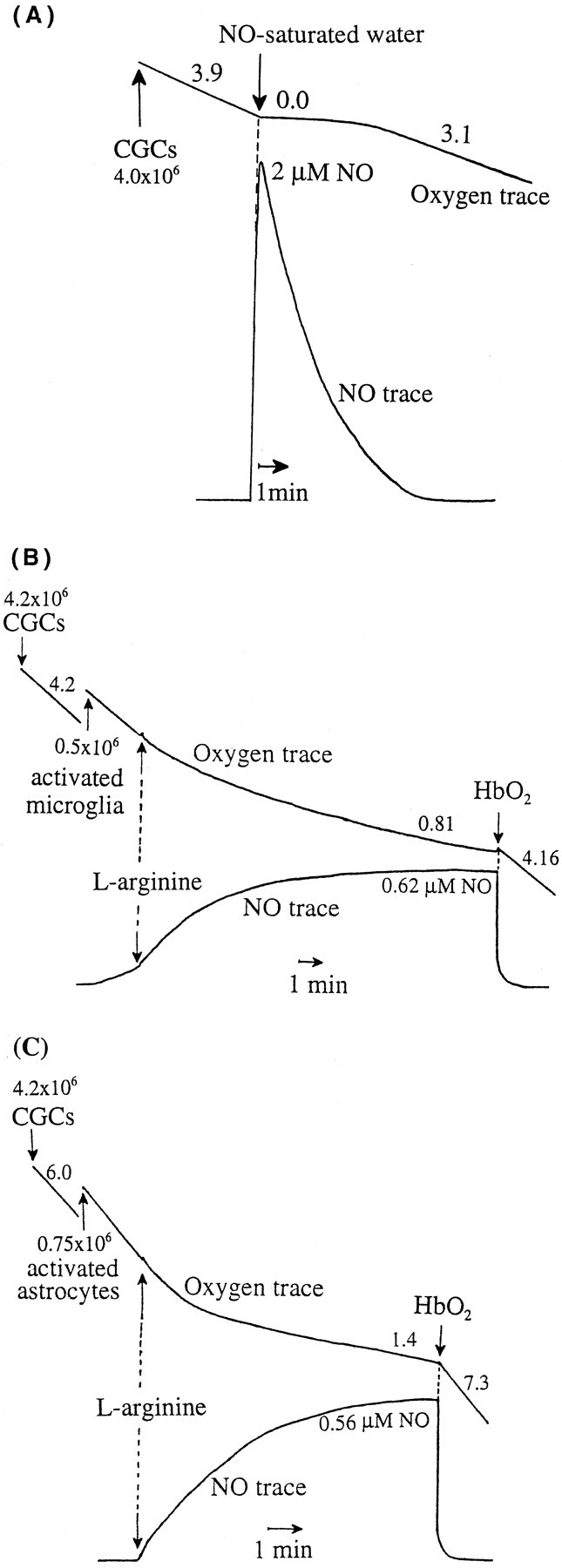
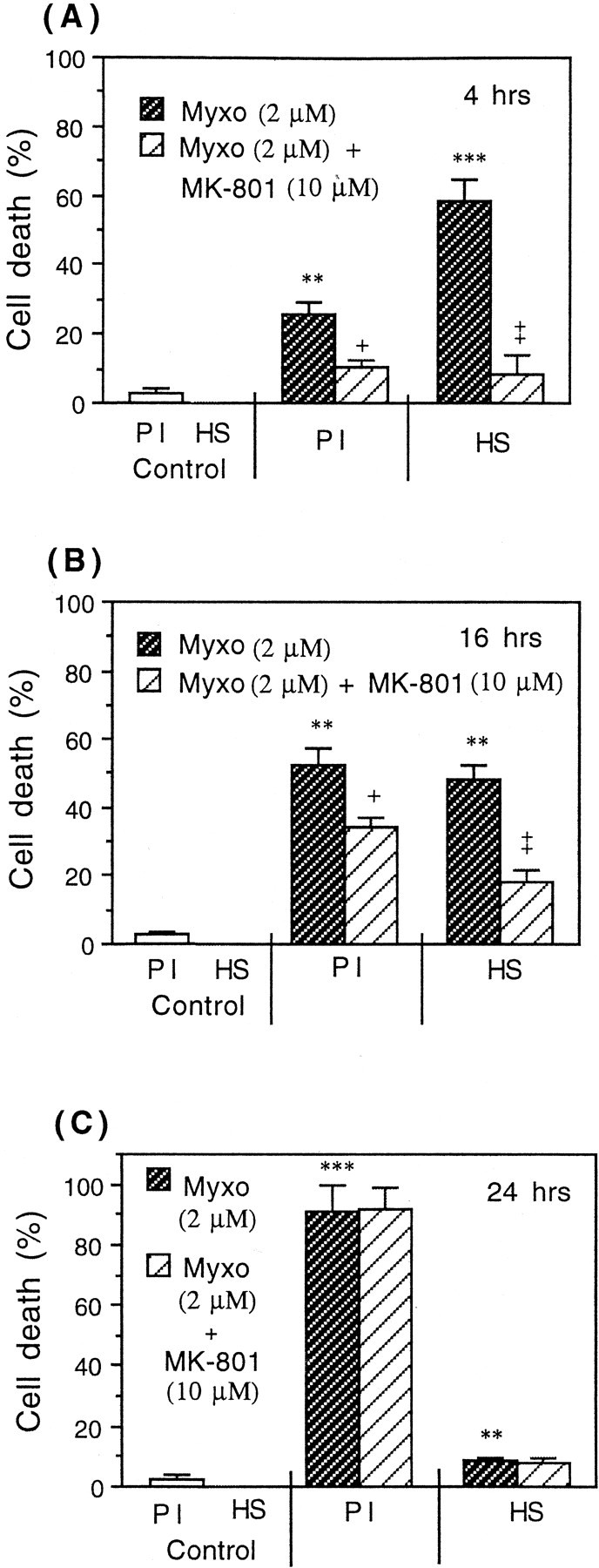
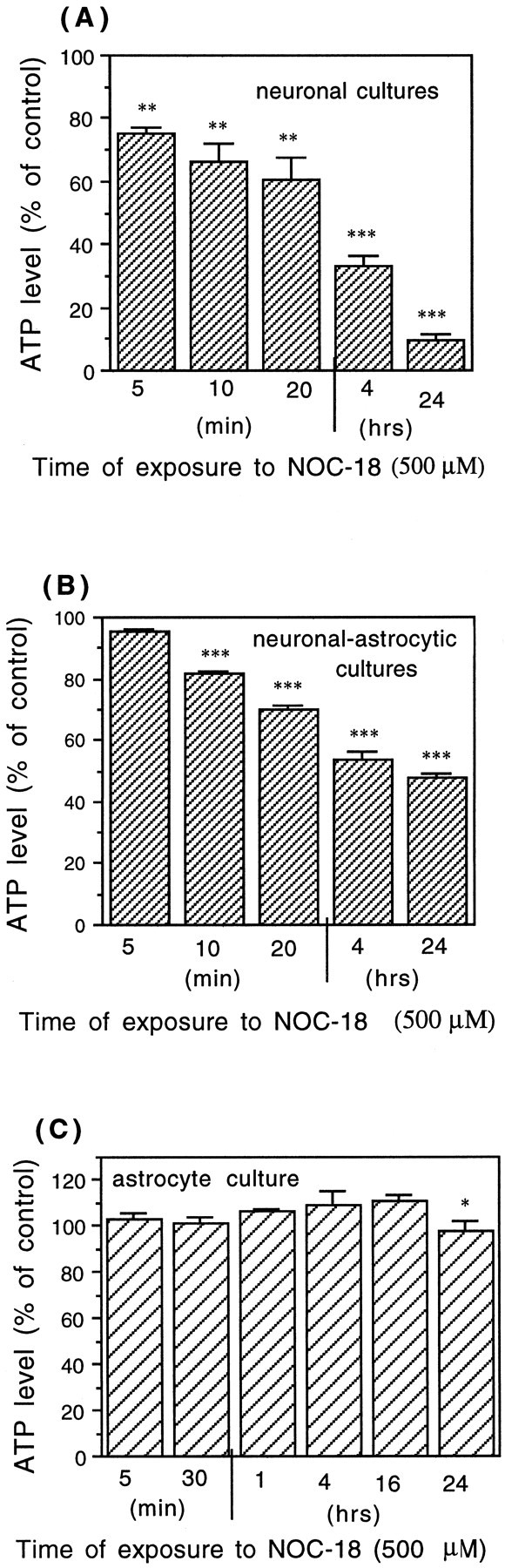
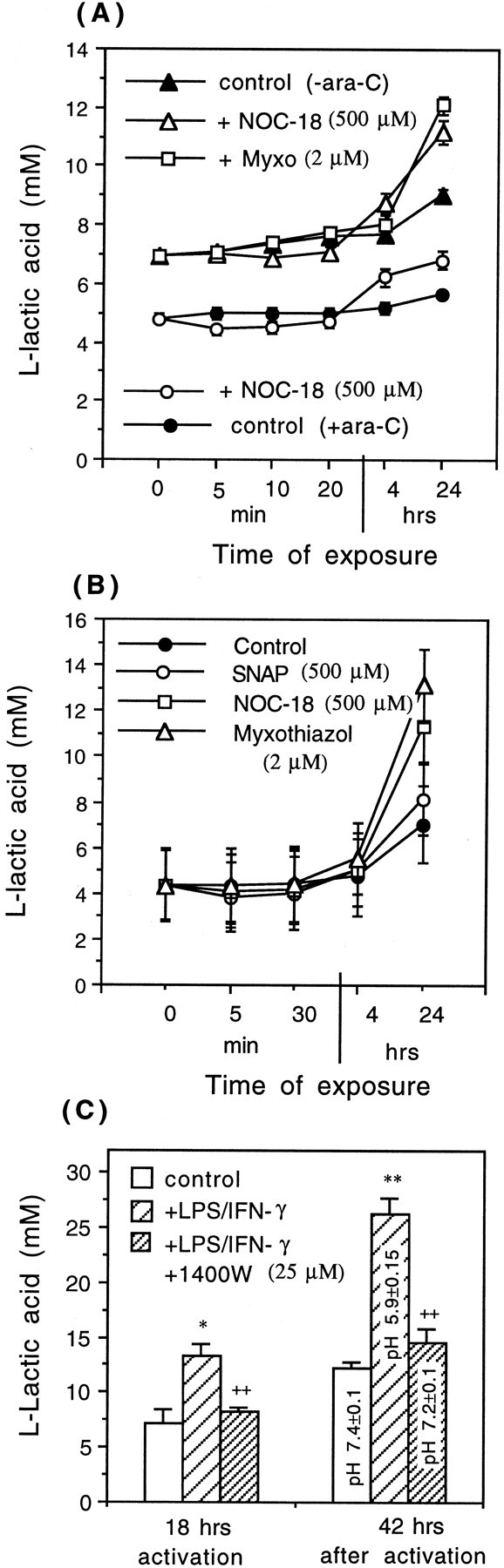
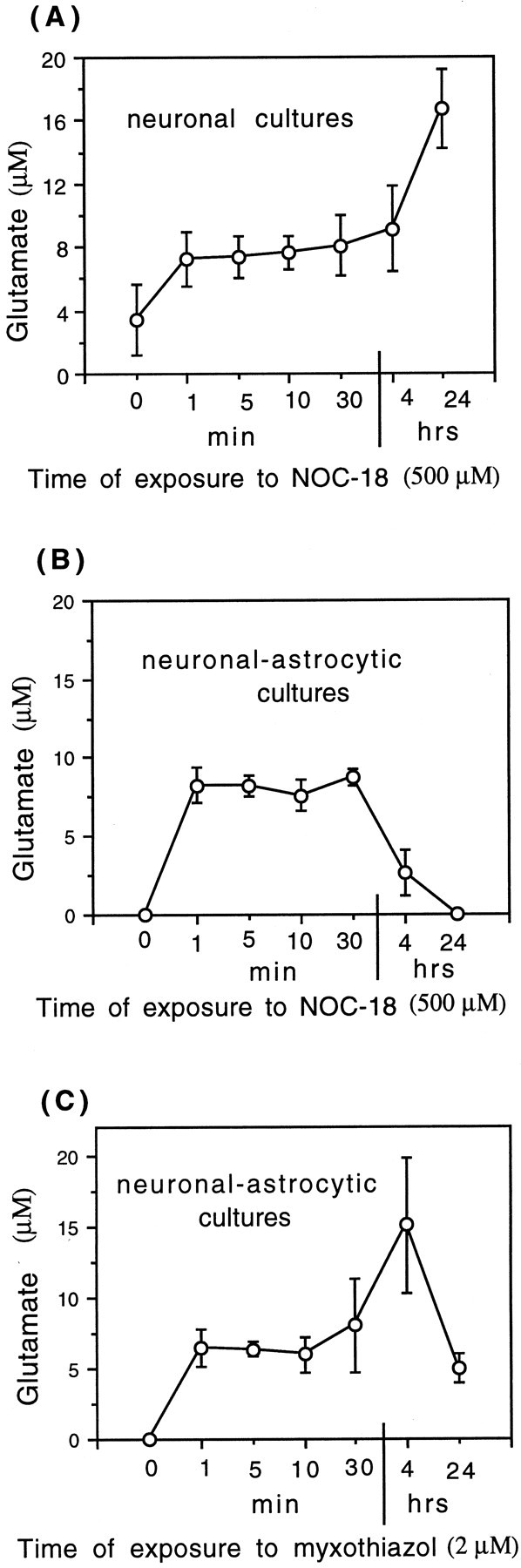
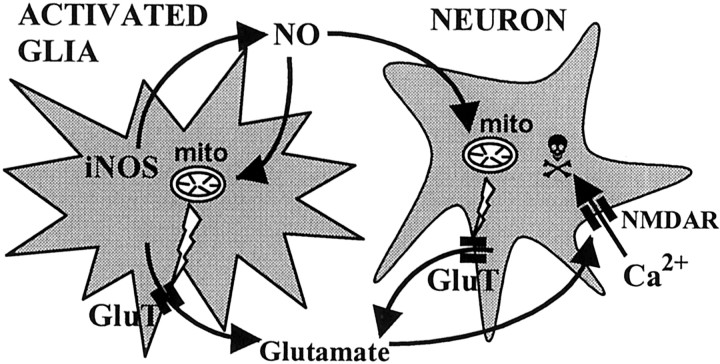
Glia undergo inflammatory activation in most CNS pathologies and are capable of killing cocultured neurons. We investigated the mechanisms of this inflammatory neurodegeneration using a mixed culture of neurons, microglia, and astrocytes, either when the astrocytes were activated directly with lipopolysaccharide (LPS) and interferon-gamma (IFN-gamma) or LPS/IFN-gamma-activated microglia were added to mixed neuronal cultures. In either case, activated glia caused 75-100% necrotic cell death within 48 hr, which was completely prevented by inhibitors of inducible nitric oxide synthase (iNOS) (aminoguanidine or 1400W). Activated astrocytes or microglia produced nitric oxide (NO) (steady-state level approximately 0.5 microm), which immediately inhibited the cellular respiration of cocultured neurons, as did authentic NO. NO donors also decreased ATP levels and stimulated lactate production by neurons, consistent with NO-induced respiratory inhibition. NO donors or a specific respiratory inhibitor caused rapid (<1 min) release of glutamate from neuronal and neuronal-astrocytic cultures and subsequent neuronal death that was blocked by an antagonist of NMDA receptor (MK-801). MK-801 also blocked neuronal death induced by activated glia. High oxygen also prevented NO-induced neuronal death, consistent with death being induced by NO inhibition of cytochrome c oxidation in competition with oxygen. Thus activated glia kill neurons via NO from iNOS, which inhibits neuronal respiration resulting in glutamate release and subsequent excitotoxicity. This may contribute to neuronal cell death in inflammatory, infectious, ischemic, and neurodegenerative diseases.
Figures
References
-
- Bal A, Bachelot T, Savasta M, Manier M, Verna JM, Benabid AL, Feuerstein C. Evidence for dopamine D2 receptor mRNA expression by striatal astrocytes in culture: in situ hybridization and polymerase chain reaction studies. Mol Brain Res. 1994;23:204–212. - PubMed
-
- Bal-Price A, Brown GC. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J Neurochem. 2000;75:1455–1464. - PubMed
-
- Banati RB, Gehramann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. - PubMed
-
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. - PubMed
-
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources