

Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3
- PMID: 11524702
- PMCID: PMC1226054
- DOI: 10.1086/323610
Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3
Erratum in
- Am J Hum Genet 2001 Nov;69(5):1160
Abstract
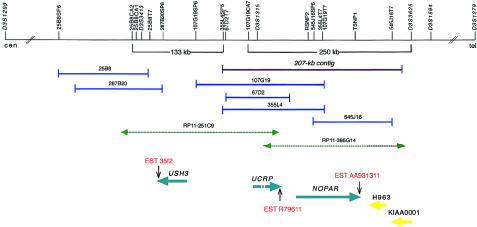
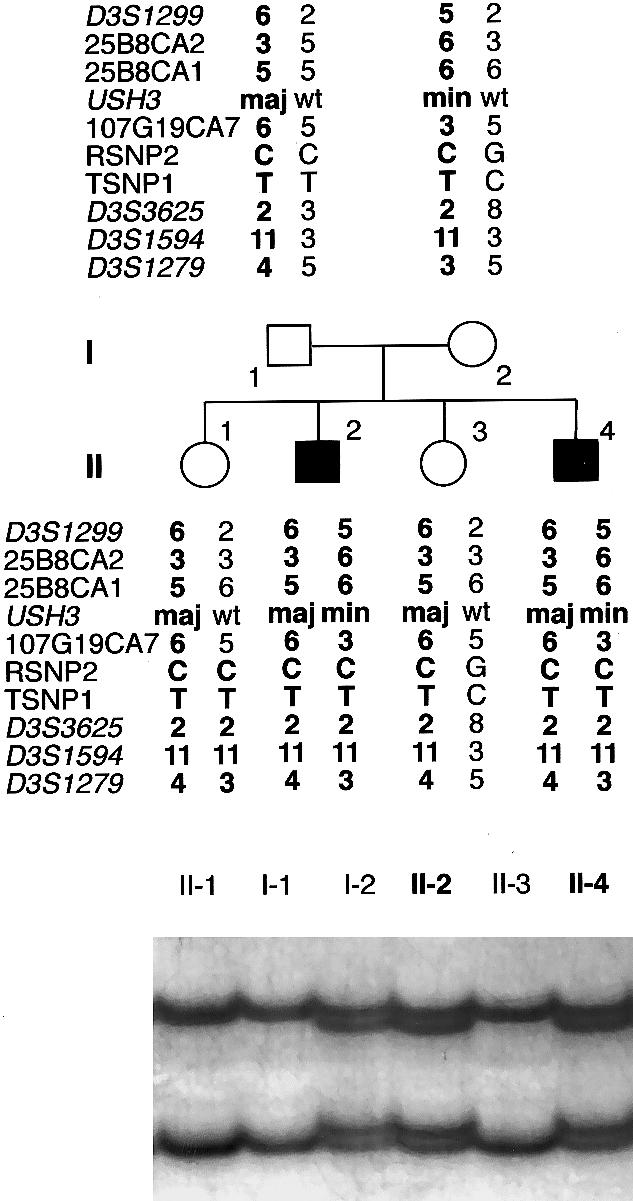
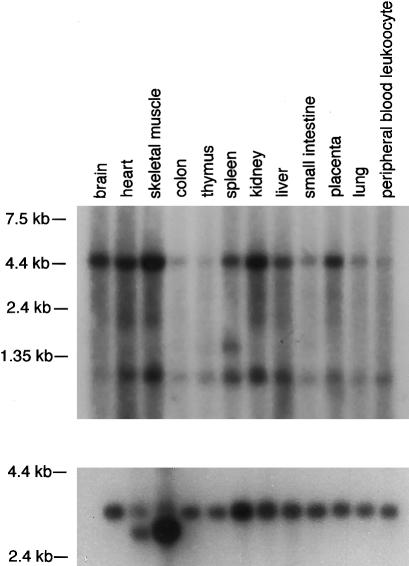
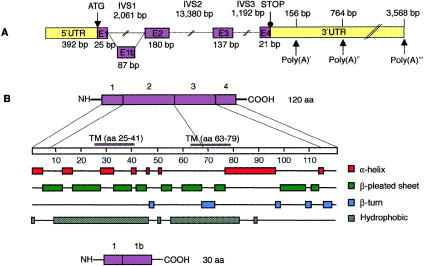
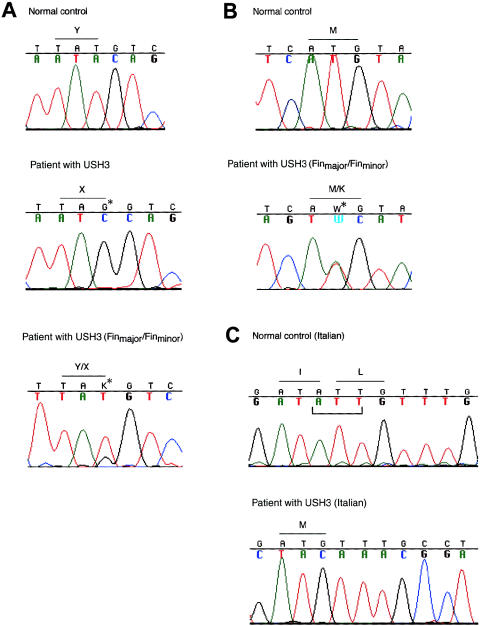
Usher syndrome type 3 (USH3) is an autosomal recessive disorder characterized by progressive hearing loss, severe retinal degeneration, and variably present vestibular dysfunction, assigned to 3q21-q25. Here, we report on the positional cloning of the USH3 gene. By haplotype and linkage-disequilibrium analyses in Finnish carriers of a putative founder mutation, the critical region was narrowed to 250 kb, of which we sequenced, assembled, and annotated 207 kb. Two novel genes-NOPAR and UCRP-and one previously identified gene-H963-were excluded as USH3, on the basis of mutational analysis. USH3, the candidate gene that we identified, encodes a 120-amino-acid protein. Fifty-two Finnish patients were homozygous for a termination mutation, Y100X; patients in two Finnish families were compound heterozygous for Y100X and for a missense mutation, M44K, whereas patients in an Italian family were homozygous for a 3-bp deletion leading to an amino acid deletion and substitution. USH3 has two predicted transmembrane domains, and it shows no homology to known genes. As revealed by northern blotting and reverse-transcriptase PCR, it is expressed in many tissues, including the retina.
Figures
References
Electronic-Database Information
-
- Baylor College of Medicine Search Launcher, http://searchlauncher.bcm.tmc.edu/ (for MatInspector/TRANSFAC and Neural Network Promoter Input programs)
-
- Celera Publication Site, http://publication.celera.com/ (for Assembled and Annotated Mouse Genome)
-
- Expressed Sequence Tags Database, http://www.ncbi.nlm.nih.gov/dbEST/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for 207-kb contig sequence [accession number AF388363], BAC clones RP11-385G14 [accession number AC011103] and RP11-251C9 [accession number AC020636], human NOPAR coding sequence [accession number AF388364], human NOPAR2 coding sequence [accession number AF388365], human UCRP putative pseudogene sequence [accession number AF388367], human HOPA coding sequence [accession number AF132033], human H963 coding sequence [accession number AF002986], human TRAP230 mRNA [accession number NM_005120], human KIAA0001 coding sequence [accession number NM_014879], EST 35f2 sequence [accession number W27577], and cDNA of USH3 [accession number AF388366] and of a splice variant, USH3 isoform b [accession number AF388368], of USH3)
References
-
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP (2001) The mouse Ames Waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet 27:99–102 - PubMed
-
- Bassam BJ, Caetano-Anolles G, Gresshoff PM (1991) Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 196:80–83 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
