

Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences
- PMID: 11532935
- PMCID: PMC125603
- DOI: 10.1093/emboj/20.17.4704
Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences
Abstract
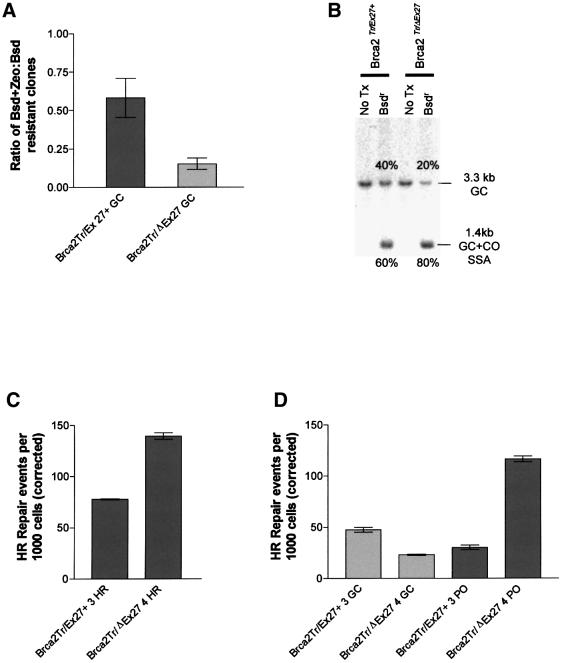
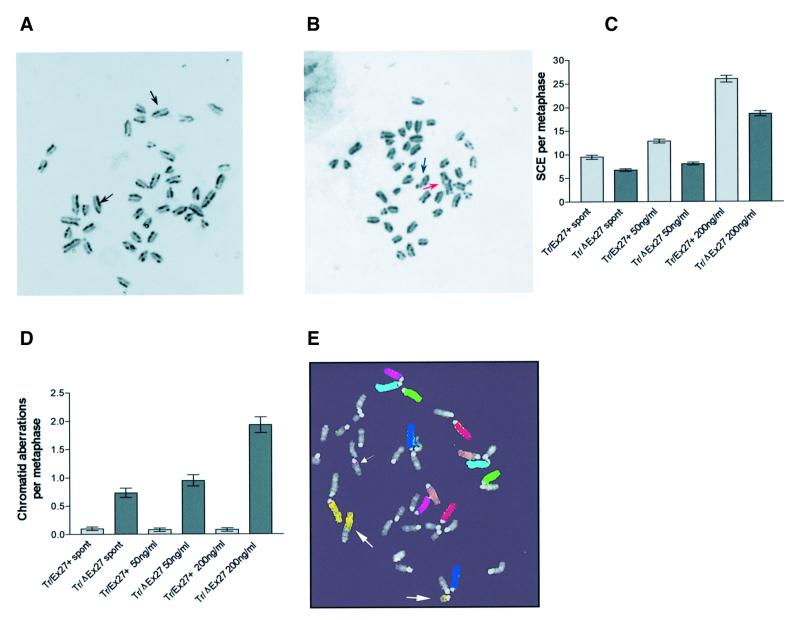
Mutation of BRCA2 causes familial early onset breast and ovarian cancer. BRCA2 has been suggested to be important for the maintenance of genome integrity and to have a role in DNA repair by homology- directed double-strand break (DSB) repair. By studying the repair of a specific induced chromosomal DSB we show that loss of Brca2 leads to a substantial increase in error-prone repair by homology-directed single-strand annealing and a reduction in DSB repair by conservative gene conversion. These data demonstrate that loss of Brca2 causes misrepair of chromosomal DSBs occurring between repeated sequences by stimulating use of an error-prone homologous recombination pathway. Furthermore, loss of Brca2 causes a large increase in genome-wide error-prone repair of both spontaneous DNA damage and mitomycin C-induced DNA cross-links at the expense of error-free repair by sister chromatid recombination. This provides insight into the mechanisms that induce genome instability in tumour cells lacking BRCA2.
Figures
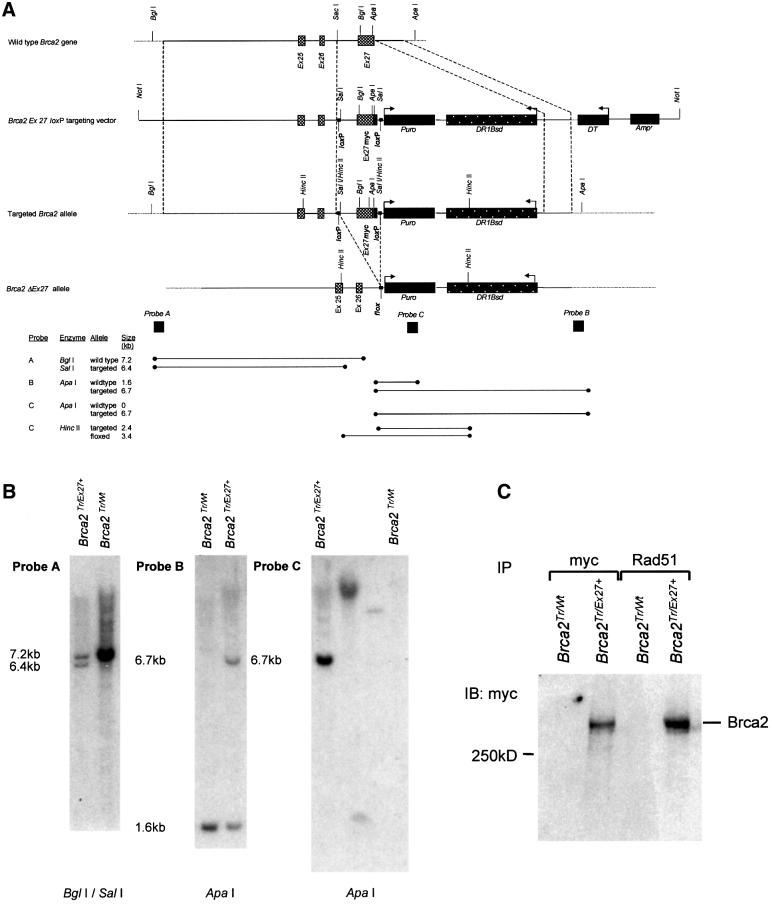
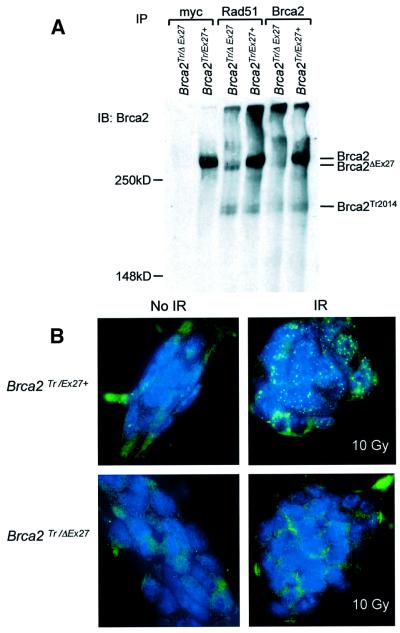
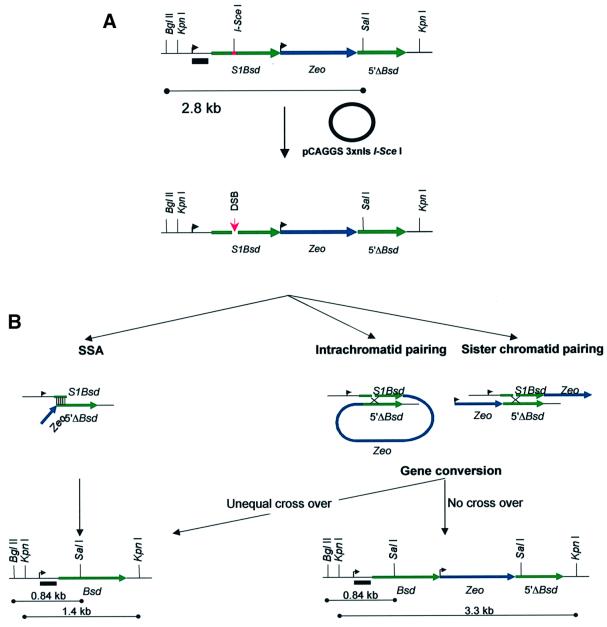
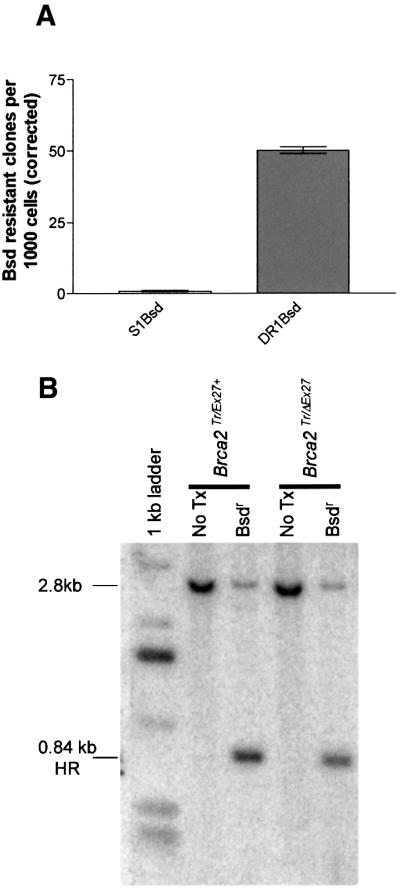
References
-
- Ban S., Shinohara,T., Hirai,Y., Moritaku,Y., Cologne,J.B. and MacPhee,D.G. (2001) Chromosomal instability in BRCA1- or BRCA2-defective human cancer cells detected by spontaneous micronucleus assay. Mutat. Res., 474, 15–23. - PubMed
-
- Baumann P. and West,S.C. (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci., 23, 247–251. - PubMed
-
- Bertwistle D. and Ashworth,A. (1998) Functions of the BRCA1 and BRCA2 genes. Curr. Opin. Genet. Dev., 8, 14–20. - PubMed
-
- Connor F., Bertwistle,D., Mee,P.J., Ross,G.M., Swift,S., Grigorieva,E., Tybulewicz,V.L. and Ashworth,A. (1997a) Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nature Genet., 17, 423–430. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
