

SGP-1: prediction and validation of homologous genes based on sequence alignments
- PMID: 11544202
- PMCID: PMC311140
- DOI: 10.1101/gr.177401
SGP-1: prediction and validation of homologous genes based on sequence alignments
Abstract
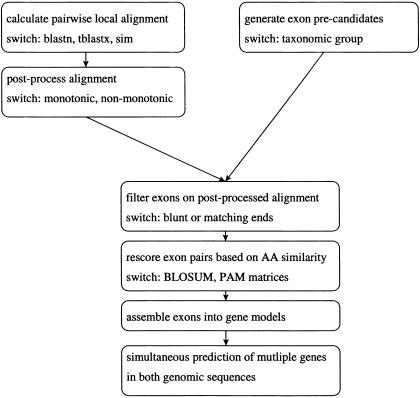
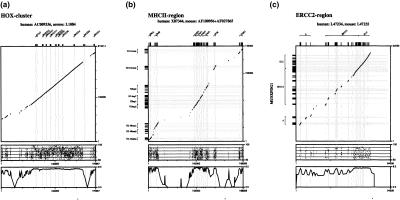
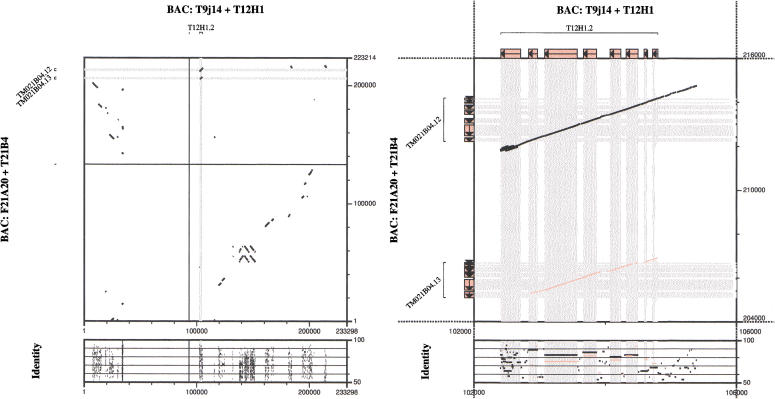
Conventional methods of gene prediction rely on the recognition of DNA-sequence signals, the coding potential or the comparison of a genomic sequence with a cDNA, EST, or protein database. Reasons for limited accuracy in many circumstances are species-specific training and the incompleteness of reference databases. Lately, comparative genome analysis has attracted increasing attention. Several analysis tools that are based on human/mouse comparisons are already available. Here, we present a program for the prediction of protein-coding genes, termed SGP-1 (Syntenic Gene Prediction), which is based on the similarity of homologous genomic sequences. In contrast to most existing tools, the accuracy of depends little on species-specific properties such as codon usage or the nucleotide distribution. may therefore be applied to nonstandard model organisms in vertebrates as well as in plants, without the need for extensive parameter training. In addition to predicting genes in large-scale genomic sequences, the program may be useful to validate gene structure annotations from databases. To this end, SGP-1 output also contains comparisons between predicted and annotated gene structures in HTML format. The program can be accessed via a Web server at http://soft.ice.mpg.de/sgp-1. The source code, written in ANSI C, is available on request from the authors.
Figures



References
-
- Abril J, Wiehe T, Guigó R. APLOT: 2D-Visualization of genome annotations. 1999. http://www1.imim.es/software/gfftools/APLOT.html http://www1.imim.es/software/gfftools/APLOT.html.
-
- Aho A, Corasick M. Efficient string matching: An aid to bibliographic search. Comm ACM. 1975;18:333–340.
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Bafna V, Huson DH. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. Menlo Park, CA: AAAI Press; 2000. The conserved exon method for gene finding; pp. 3–12. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials