

The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis
- PMID: 11551981
- PMCID: PMC2150817
- DOI: 10.1083/jcb.200105020
The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis
Abstract
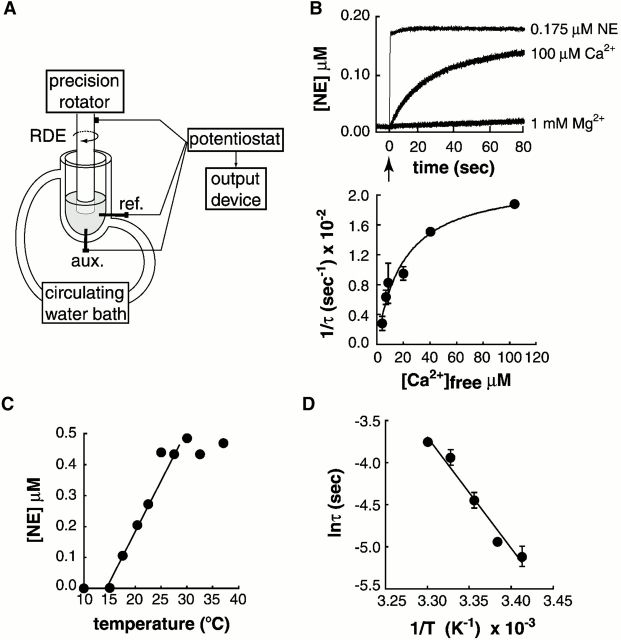
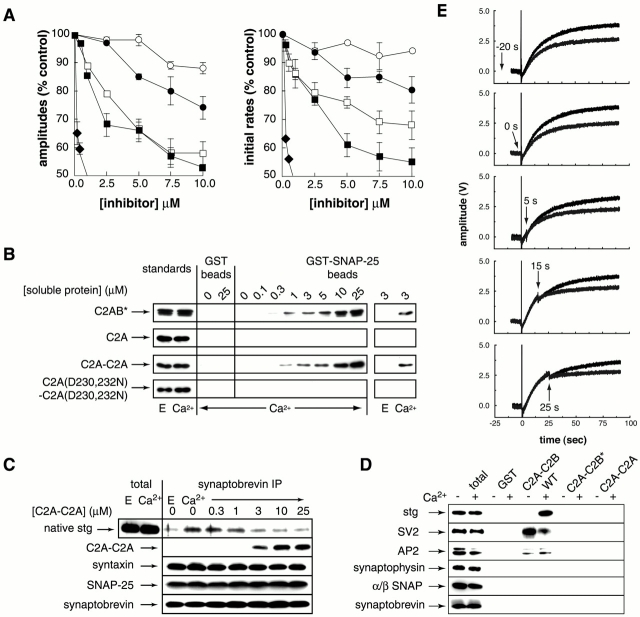
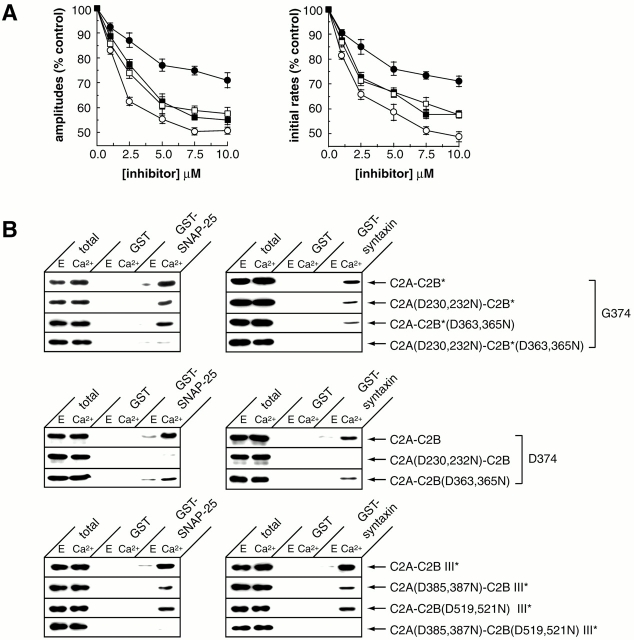
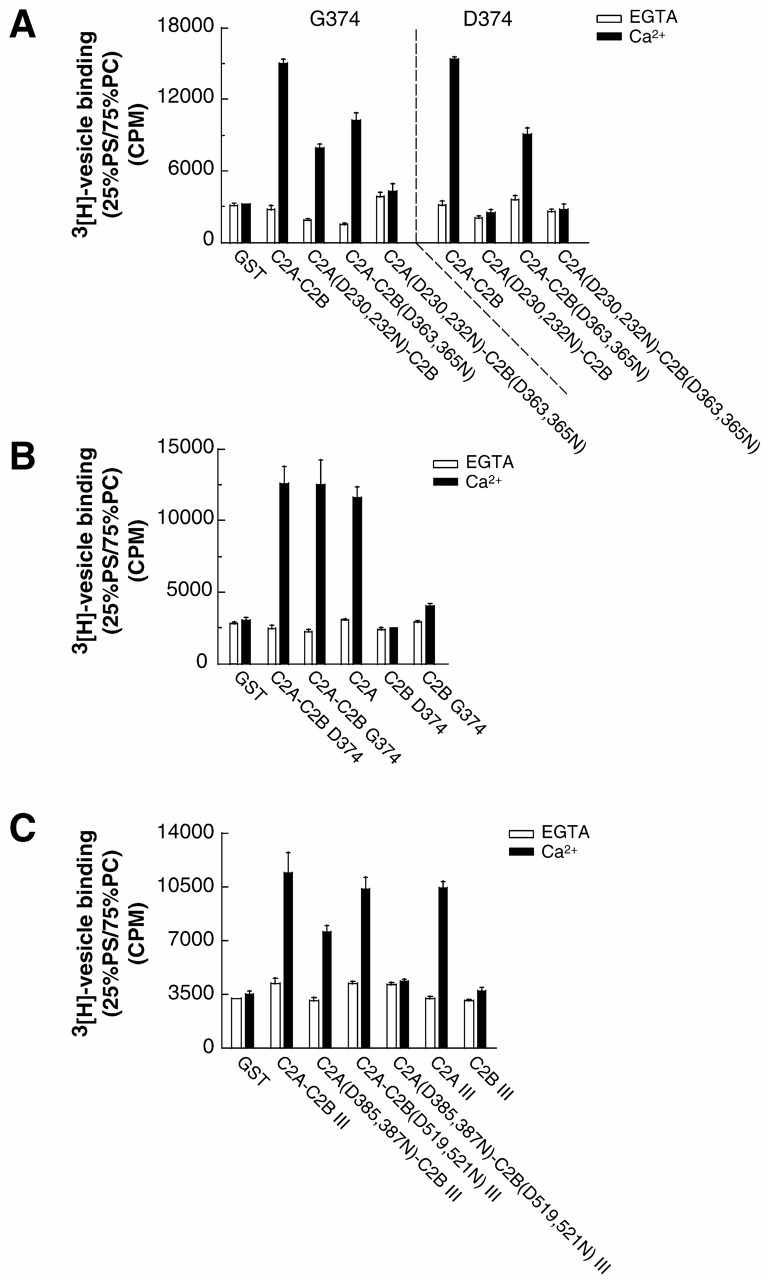
Real-time voltammetry measurements from cracked PC12 cells were used to analyze the role of synaptotagmin-SNARE interactions during Ca2+-triggered exocytosis. The isolated C2A domain of synaptotagmin I neither binds SNAREs nor inhibits norepinephrine secretion. In contrast, two C2 domains in tandem (either C2A-C2B or C2A-C2A) bind strongly to SNAREs, displace native synaptotagmin from SNARE complexes, and rapidly inhibit exocytosis. The tandem C2 domains of synaptotagmin cooperate via a novel mechanism in which the disruptive effects of Ca2+ ligand mutations in one C2 domain can be partially alleviated by the presence of an adjacent C2 domain. Complete disruption of Ca2+-triggered membrane and target membrane SNARE interactions required simultaneous neutralization of Ca2+ ligands in both C2 domains of the protein. We conclude that synaptotagmin-SNARE interactions regulate membrane fusion and that cooperation between synaptotagmin's C2 domains is crucial to its function.
Figures
References
-
- Augustine, G.J. 2001. How does calcium trigger neurotransmitter release? Curr. Opin. Neuro. 11:320–326. - PubMed
-
- Bai, J., C. Earles, J. Lewis, and E.R. Chapman. 2000. Membrane-embedded synaptotagmin penetrates cis and trans target membranes and clusters via a novel mechanism. J. Biol. Chem. 275:25427–25435. - PubMed
-
- Bark, I.C., and M.C. Wilson. 1994. Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene. 139:291–292. - PubMed
-
- Bennett, M.K., N. Calakos, and R.H. Scheller. 1992. Syntaxin—a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 257:255–259. - PubMed
-
- Brose, N.A., G. Petrenko, T.C. Südhof, and R. Jahn. 1992. Synaptotagmin: a Ca2+ sensor on the synaptic vesicle surface. Science. 256:1021–1025. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
