

Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC
- PMID: 11553773
- PMCID: PMC58692
- DOI: 10.1073/pnas.191369098
Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC
Abstract
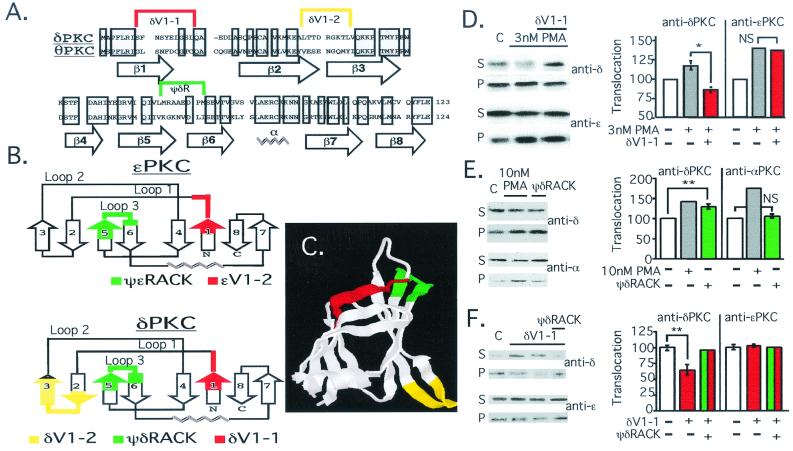
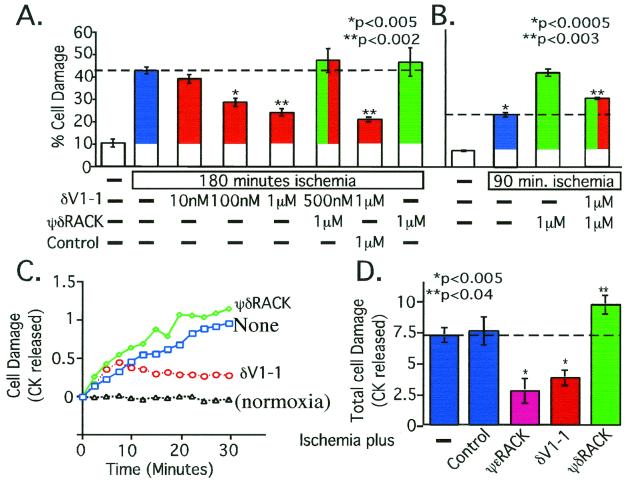
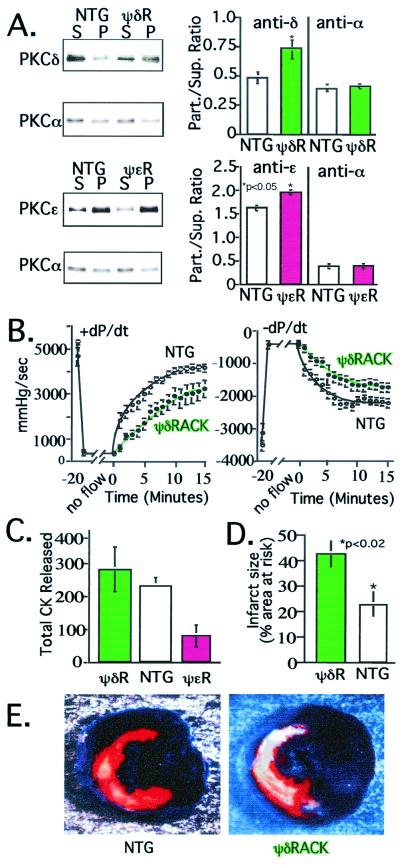
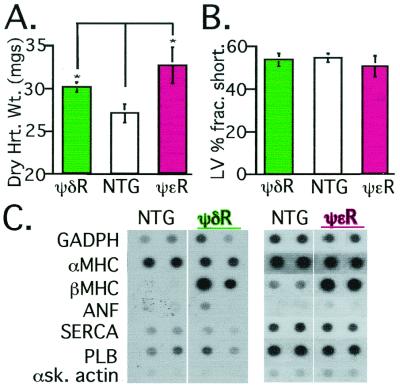
Conflicting roles for protein kinase C (PKC) isozymes in cardiac disease have been reported. Here, deltaPKC-selective activator and inhibitor peptides were designed rationally, based on molecular modeling and structural homology analyses. Together with previously identified activator and inhibitor peptides of epsilonPKC, deltaPKC peptides were used to identify cardiac functions of these isozymes. In isolated cardiomyocytes, perfused hearts, and transgenic mice, deltaPKC and epsilonPKC had opposing actions on protection from ischemia-induced damage. Specifically, activation of epsilonPKC caused cardioprotection whereas activation of deltaPKC increased damage induced by ischemia in vitro and in vivo. In contrast, deltaPKC and epsilonPKC caused identical nonpathological cardiac hypertrophy; activation of either isozyme caused nonpathological hypertrophy of the heart. These results demonstrate that two related PKC isozymes have both parallel and opposing effects in the heart, indicating the danger in the use of therapeutics with nonselective isozyme inhibitors and activators. Moreover, reduction in cardiac damage caused by ischemia by perfusion of selective regulator peptides of PKC through the coronary arteries constitutes a major step toward developing a therapeutic agent for acute cardiac ischemia.
Figures
References
-
- Brooks G, Hearse D J. Circ Res. 1996;79:627–630. - PubMed
-
- Gu X, Bishop S P. Circ Res. 1994;75:926–931. - PubMed
-
- Bowling N, Walsh R A, Song G, Estridge T, Sandusky G E, Fouts R L, Mintze K, Pickard T, Roden R, Bristow M R, et al. Circulation. 1999;99:384–391. - PubMed
-
- Jalili T, Takeishi Y, Song G, Ball N A, Howles G, Walsh R A. Am J Physiol. 1999;277:H2298–H2304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
