

7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome
- PMID: 11560960
- PMCID: PMC200927
- DOI: 10.1172/JCI12103
7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome
Abstract
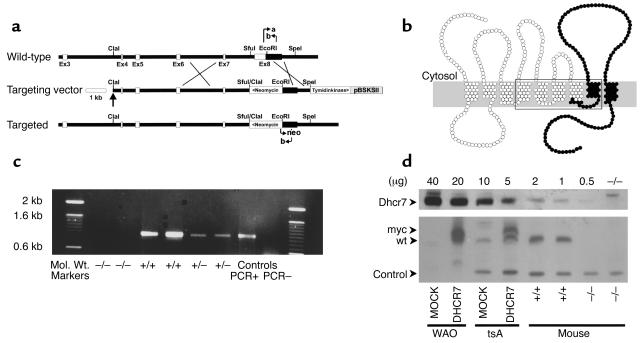
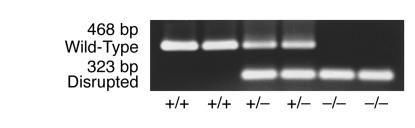
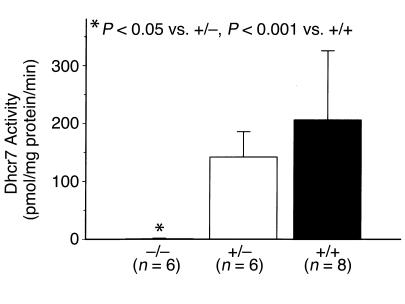
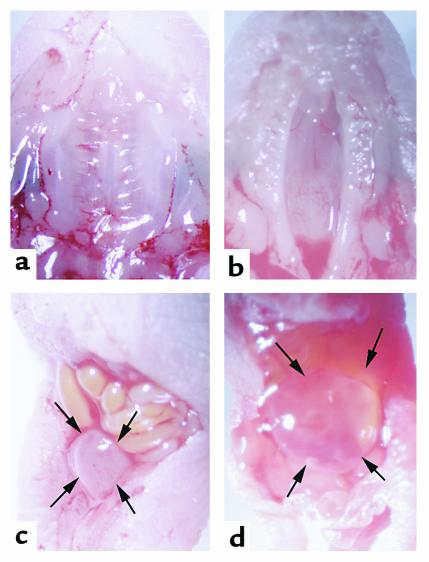
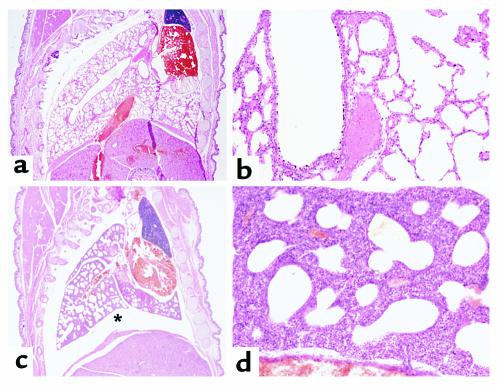
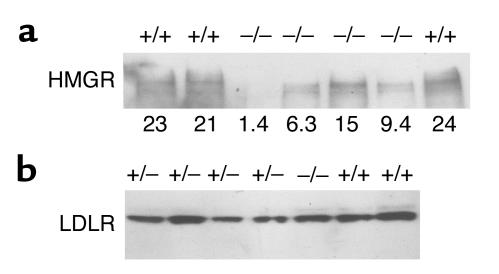
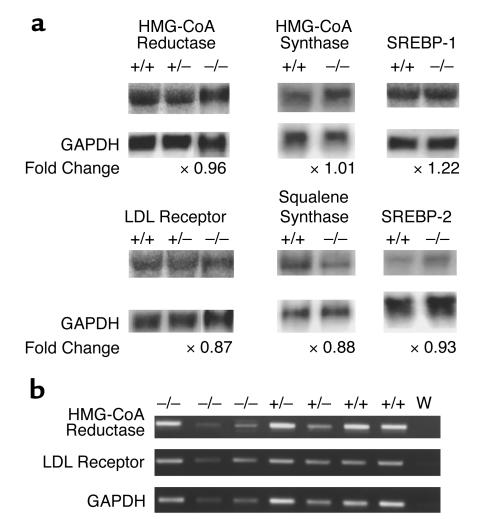
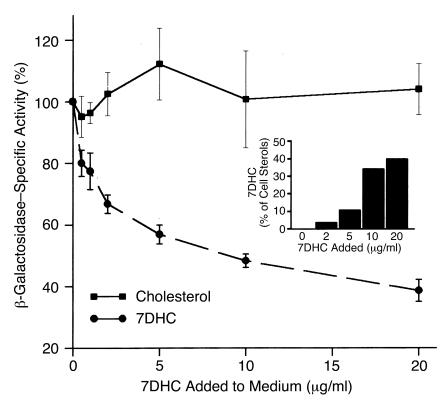
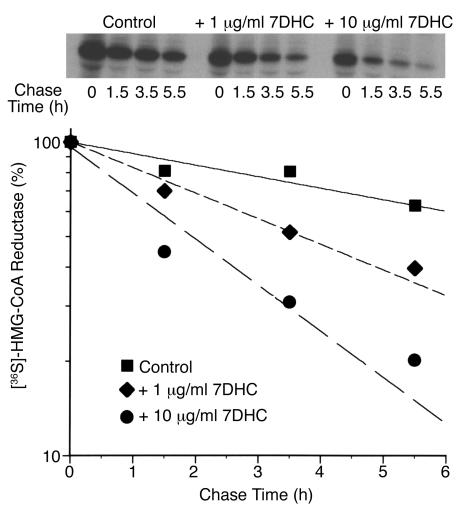
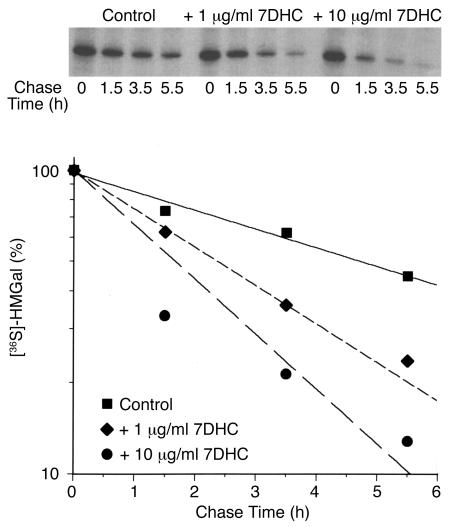
Smith-Lemli-Opitz/RSH syndrome (SLOS), a relatively common birth-defect mental-retardation syndrome, is caused by mutations in DHCR7, whose product catalyzes an obligate step in cholesterol biosynthesis, the conversion of 7-dehydrocholesterol to cholesterol. A null mutation in the murine Dhcr7 causes an identical biochemical defect to that seen in SLOS, including markedly reduced tissue cholesterol and total sterol levels, and 30- to 40-fold elevated concentrations of 7-dehydrocholesterol. Prenatal lethality was not noted, but newborn homozygotes breathed with difficulty, did not suckle, and died soon after birth with immature lungs, enlarged bladders, and, frequently, cleft palates. Despite reduced sterol concentrations in Dhcr7(-/-) mice, mRNA levels for 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-controlling enzyme for sterol biosynthesis, the LDL receptor, and SREBP-2 appeared neither elevated nor repressed. In contrast to mRNA, protein levels and activities of HMG-CoA reductase were markedly reduced. Consistent with this finding, 7-dehydrocholesterol accelerates proteolysis of HMG-CoA reductase while sparing other key proteins. These results demonstrate that in mice without Dhcr7 activity, accumulated 7-dehydrocholesterol suppresses sterol biosynthesis posttranslationally. This effect might exacerbate abnormal development in SLOS by increasing the fetal cholesterol deficiency.
Figures
Similar articles
-
Regulation of cholesterol biosynthetic pathway in patients with the Smith-Lemli-Opitz syndrome.J Inherit Metab Dis. 2000 Jul;23(5):464-74. doi: 10.1023/a:1005660130109. J Inherit Metab Dis. 2000. PMID: 10947201
-
Rapid identification of Smith-Lemli-Opitz syndrome homozygotes and heterozygotes (carriers) by measurement of deficient 7-dehydrocholesterol-delta 7-reductase activity in fibroblasts.Metabolism. 1997 Jul;46(7):844-50. doi: 10.1016/s0026-0495(97)90133-5. Metabolism. 1997. PMID: 9225842
-
7-Dehydrocholesterol down-regulates cholesterol biosynthesis in cultured Smith-Lemli-Opitz syndrome skin fibroblasts.J Lipid Res. 1998 Mar;39(3):647-57. J Lipid Res. 1998. PMID: 9548596
-
Abnormal cholesterol biosynthesis in sitosterolaemia and the Smith-Lemli-Opitz syndrome.J Inherit Metab Dis. 1996;19(4):391-400. doi: 10.1007/BF01799100. J Inherit Metab Dis. 1996. PMID: 8884563 Review.
-
RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis.Mol Genet Metab. 2000 Sep-Oct;71(1-2):163-74. doi: 10.1006/mgme.2000.3069. Mol Genet Metab. 2000. PMID: 11001807 Review.
Cited by
-
Atypical antipsychotics alter cholesterol and fatty acid metabolism in vitro.J Lipid Res. 2013 Feb;54(2):310-24. doi: 10.1194/jlr.M026948. Epub 2012 Nov 21. J Lipid Res. 2013. PMID: 23175778 Free PMC article.
-
Molecular consequences of altered neuronal cholesterol biosynthesis.J Neurosci Res. 2009 Mar;87(4):866-75. doi: 10.1002/jnr.21917. J Neurosci Res. 2009. PMID: 18951487 Free PMC article.
-
7-Dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome.Biochim Biophys Acta. 2012 Jun;1821(6):877-83. doi: 10.1016/j.bbalip.2012.03.001. Epub 2012 Mar 9. Biochim Biophys Acta. 2012. PMID: 22425966 Free PMC article.
-
Exploring Recent Developments in the Manifestation, Diagnosis, and Treatment of Patients with Smith-Lemli-Opitz Syndrome: From Molecular Pathways to Clinical Innovations.Int J Mol Sci. 2025 Jul 11;26(14):6672. doi: 10.3390/ijms26146672. Int J Mol Sci. 2025. PMID: 40724921 Free PMC article. Review.
-
7-dehydrocholesterol efficiently supports Ret signaling in a mouse model of Smith-Opitz-Lemli syndrome.Sci Rep. 2016 Jun 23;6:28534. doi: 10.1038/srep28534. Sci Rep. 2016. PMID: 27334845 Free PMC article.
References
-
- Hoffmann G, et al. Mevalonic aciduria: an inborn error of cholesterol and nonsterol isoprene biosynthesis. N Engl J Med. 1986;314:1610–1614. - PubMed
-
- Clayton P, Mills K, Keeling J, FitzPatrick D. Desmosterolosis: a new inborn error of cholesterol biosynthesis. Lancet. 1996;348:404. - PubMed
-
- Kelley RI, et al. Abnormal sterol metabolism in patients with Conradi-Hunermann-Happle syndrome and sporadic lethal chondrodysplasia punctata. Am J Med Genet. 1999;83:213–219. - PubMed
-
- Braverman N, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-Δ8,Δ7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999;22:291–294. - PubMed
-
- König A, Happle R, Bornholdt D, Engel H, Grzeschik K-H. Mutations in the NSDHL gene, encoding a 3β-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339–346. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
