

Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand
- PMID: 11567045
- PMCID: PMC6762892
- DOI: 10.1523/JNEUROSCI.21-19-07551.2001
Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand
Abstract
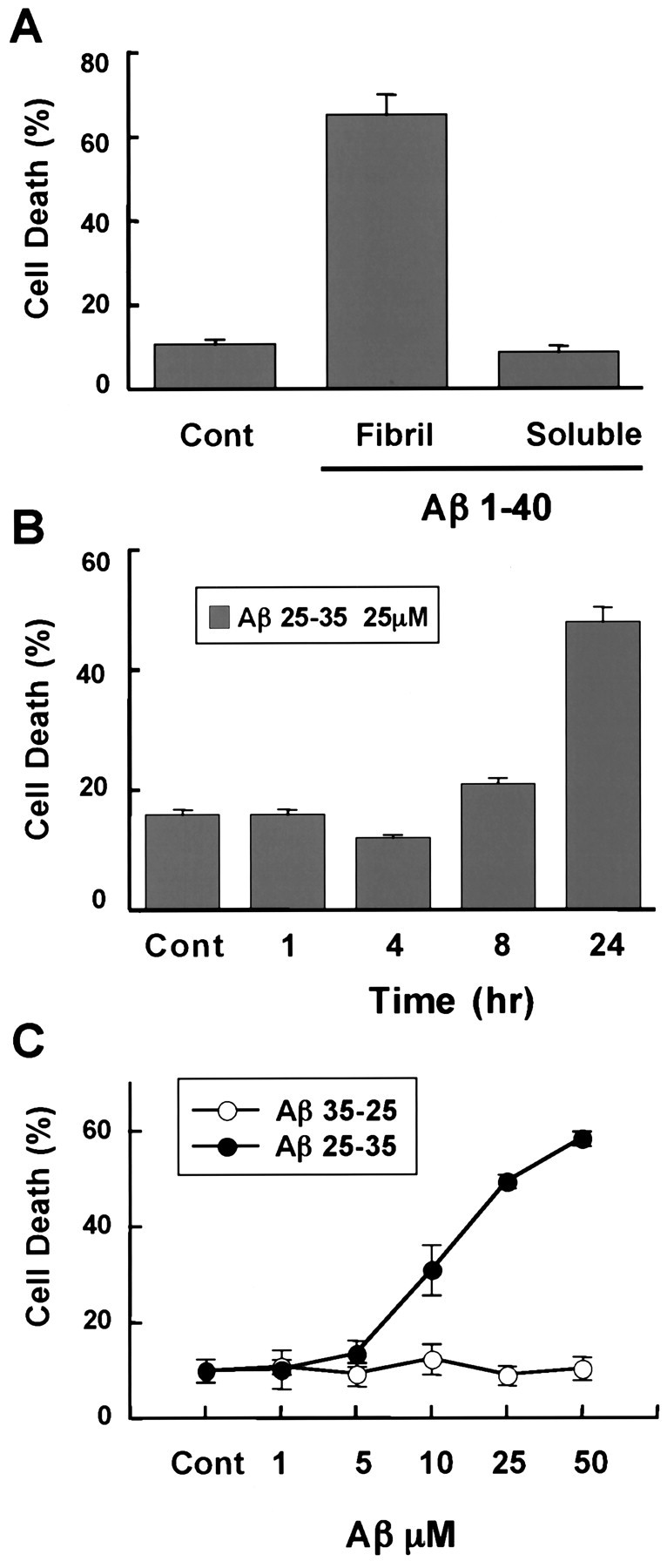
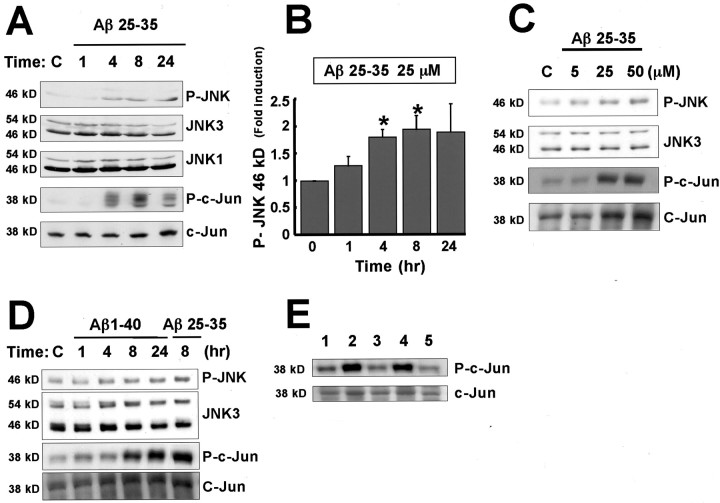
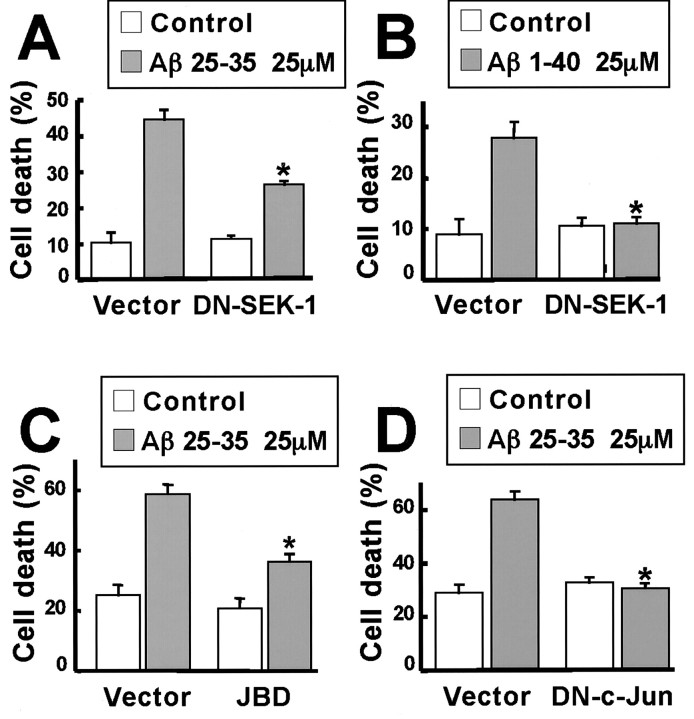
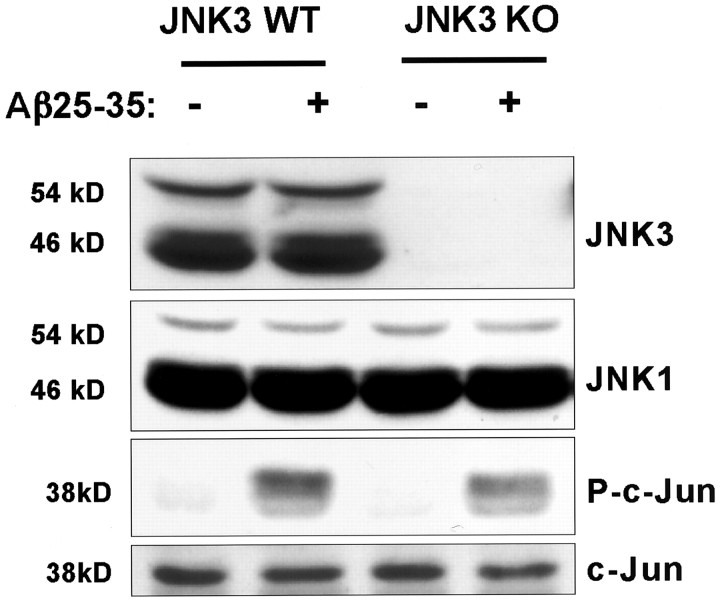
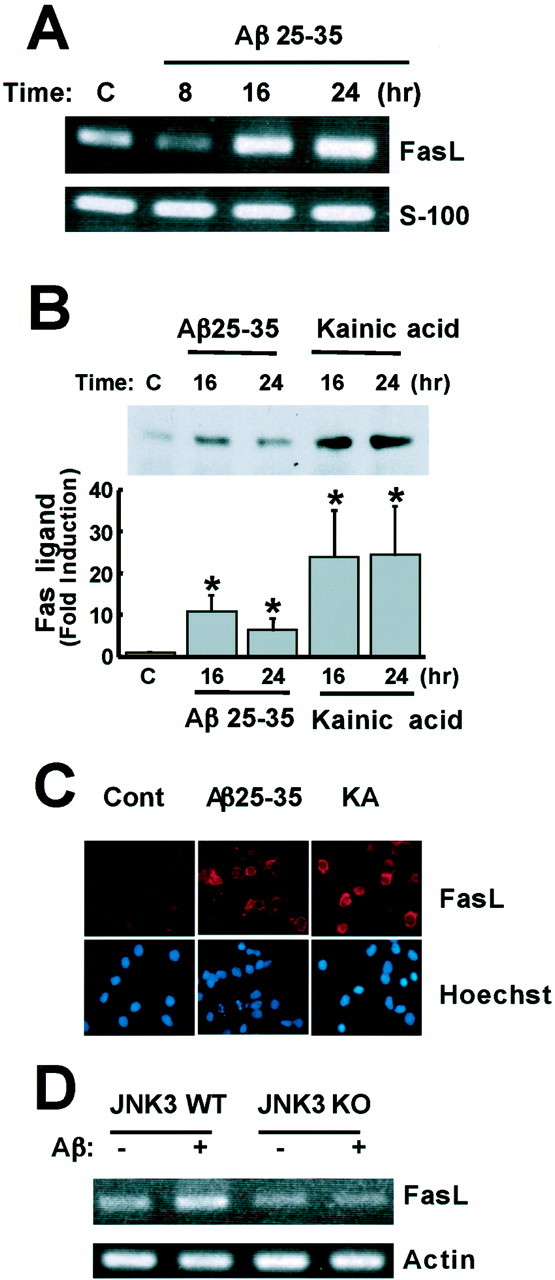
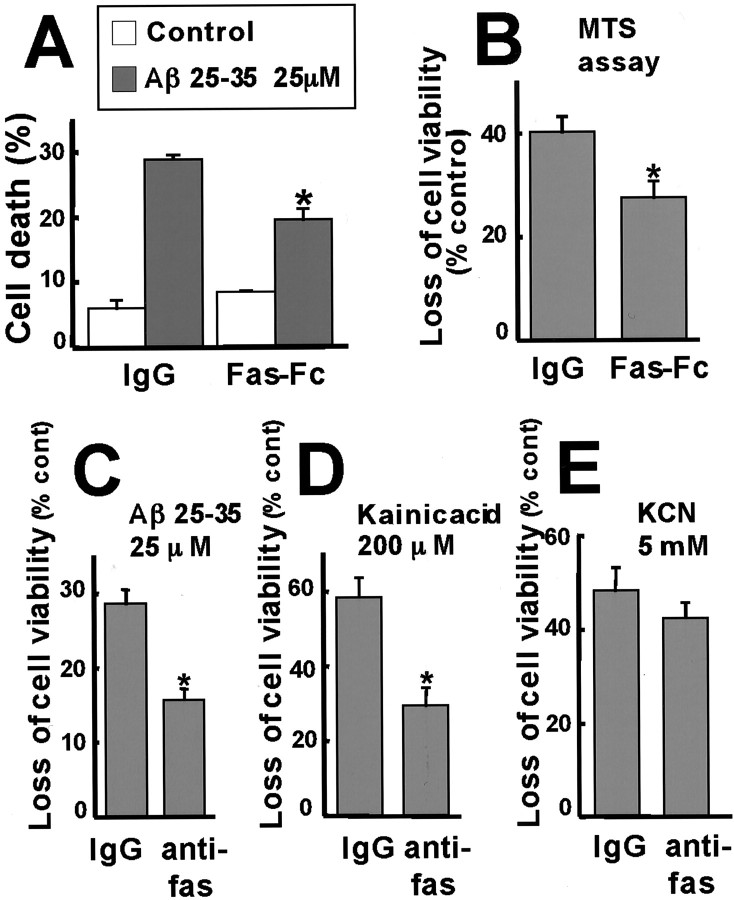
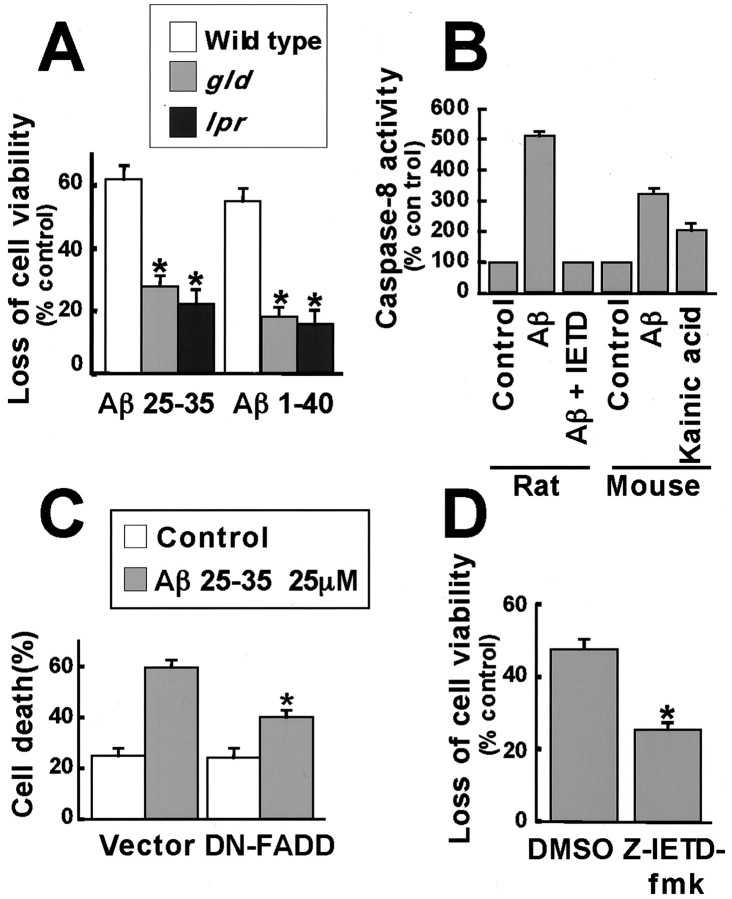
Elevated levels of beta-Amyloid (Abeta) are present in the brains of individuals with either the sporadic or familial form of Alzheimer's disease (AD), and the deposition of Abeta within the senile plaques that are a hallmark of AD is thought to be a primary cause of the cognitive dysfunction that occurs in AD. Recent evidence suggests that Abeta induces neuronal apoptosis in the brain and in primary neuronal cultures, and that this Abeta-induced neuronal death may be responsible in part for the cognitive decline found in AD patients. In this study we have characterized one mechanism by which Abeta induces neuronal death. We found that in cortical neurons exposed to Abeta, activated c-Jun N-terminal kinase (JNK) is required for the phosphorylation and activation of the c-Jun transcription factor, which in turn stimulates the transcription of several key target genes, including the death inducer Fas ligand. The binding of Fas ligand to its receptor Fas then induces a cascade of events that lead to caspase activation and ultimately cell death. By analyzing the effects of mutations in each of the components of the JNK-c-Jun-Fas ligand-Fas pathway, we demonstrate that this pathway plays a critical role in mediating Abeta-induced death of cultured neurons. These findings raise the possibility that the JNK pathway may also contribute to Abeta-dependent death in AD patients.
Figures

References
-
- Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease. Exp Neurol. 1994;125:286–295. - PubMed
-
- Behrens A, Sibilis M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. - PubMed
-
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phospholylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous