

A novel approach for assessing macromolecular complexes combining soft-docking calculations with NMR data
- PMID: 11567104
- PMCID: PMC2374225
- DOI: 10.1110/ps.07501
A novel approach for assessing macromolecular complexes combining soft-docking calculations with NMR data
Abstract
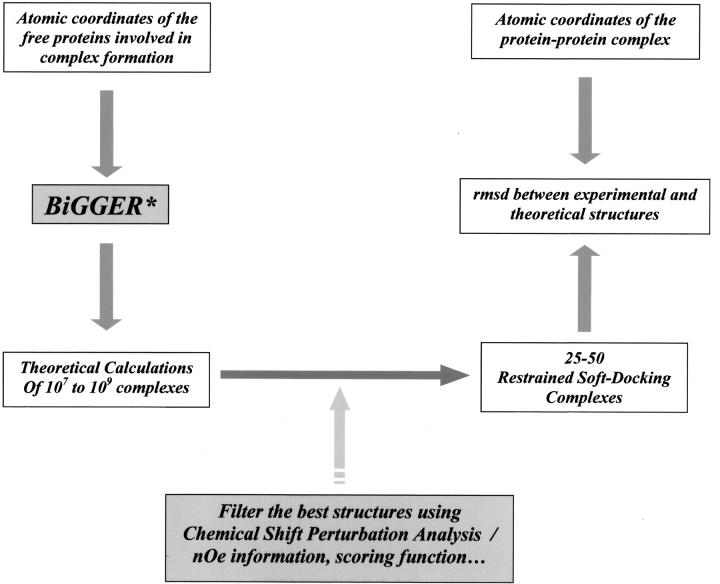
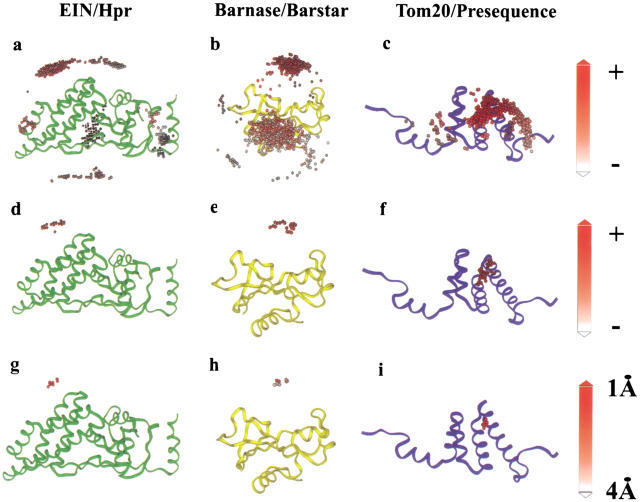
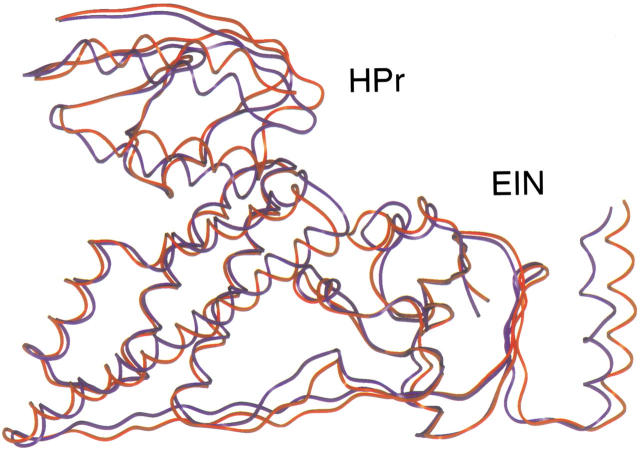
We present a novel and efficient approach for assessing protein-protein complex formation, which combines ab initio docking calculations performed with the protein docking algorithm BiGGER and chemical shift perturbation data collected with heteronuclear single quantum coherence (HSQC) or TROSY nuclear magnetic resonance (NMR) spectroscopy. This method, termed "restrained soft-docking," is validated for several known protein complexes. These data demonstrate that restrained soft-docking extends the size limitations of NMR spectroscopy and provides an alternative method for investigating macromolecular protein complexes that requires less experimental time, effort, and resources. The potential utility of this novel NMR and simulated docking approach in current structural genomic initiatives is discussed.
Figures
Similar articles
-
Docking of protein-protein complexes on the basis of highly ambiguous intermolecular distance restraints derived from 1H/15N chemical shift mapping and backbone 15N-1H residual dipolar couplings using conjoined rigid body/torsion angle dynamics.J Am Chem Soc. 2003 Mar 12;125(10):2902-12. doi: 10.1021/ja028893d. J Am Chem Soc. 2003. PMID: 12617657
-
Filtering and selection of structural models: combining docking and NMR.Proteins. 2003 Oct 1;53(1):18-32. doi: 10.1002/prot.10439. Proteins. 2003. PMID: 12945046
-
Structures of protein-protein complexes are docked using only NMR restraints from residual dipolar coupling and chemical shift perturbations.J Am Chem Soc. 2002 Mar 13;124(10):2104-5. doi: 10.1021/ja017242z. J Am Chem Soc. 2002. PMID: 11878950
-
Three-dimensional structures of protein-protein complexes in the E. coli PTS.J Mol Microbiol Biotechnol. 2001 Jul;3(3):347-54. J Mol Microbiol Biotechnol. 2001. PMID: 11361064 Review.
-
Three-dimensional structures of the central regulatory proteins of the bacterial phosphotransferase system, HPr and enzyme IIAglc.J Cell Biochem. 1993 Jan;51(1):75-82. doi: 10.1002/jcb.240510114. J Cell Biochem. 1993. PMID: 8432747 Review.
Cited by
-
Predicting Protein-Protein Interactions Using BiGGER: Case Studies.Molecules. 2016 Aug 9;21(8):1037. doi: 10.3390/molecules21081037. Molecules. 2016. PMID: 27517887 Free PMC article.
-
High-resolution structure determination of the CylR2 homodimer using paramagnetic relaxation enhancement and structure-based prediction of molecular alignment.J Biomol NMR. 2008 Jan;40(1):1-13. doi: 10.1007/s10858-007-9204-4. Epub 2007 Nov 20. J Biomol NMR. 2008. PMID: 18026911 Free PMC article.
-
Synergistic applications of MD and NMR for the study of biological systems.J Biomed Biotechnol. 2012;2012:254208. doi: 10.1155/2012/254208. Epub 2012 Jan 26. J Biomed Biotechnol. 2012. PMID: 22319241 Free PMC article. Review.
-
Efficient docking of peptides to proteins without prior knowledge of the binding site.Protein Sci. 2002 Jul;11(7):1729-37. doi: 10.1110/ps.0202302. Protein Sci. 2002. PMID: 12070326 Free PMC article.
-
Structural bioinformatics and protein docking analysis of the molecular chaperone-kinase interactions: towards allosteric inhibition of protein kinases by targeting the hsp90-cdc37 chaperone machinery.Pharmaceuticals (Basel). 2013 Nov 11;6(11):1407-28. doi: 10.3390/ph6111407. Pharmaceuticals (Basel). 2013. PMID: 24287464 Free PMC article.
References
-
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. - PubMed
-
- Abe, Y., Shodai, T., Muto, T., Mihara, K., Torii, H., Nishikawa, S., Endo, T., and Kohda, D. 2000. Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100 551–560. - PubMed
-
- Bacon, D.J. and Moult, J. 1992. Docking by least-squares fitting of molecular surface patterns. J. Mol. Biol. 225 849–858. - PubMed
-
- Bolon, P.J., Al-Hashimi, H.M., and Prestegard, J.H. 1999. Residual dipolar coupling derived orientational constraints on ligand geometry in a 53 kDa protein-ligand complex. J. Mol. Biol. 293 107–115. - PubMed
-
- Buckle, A.M., Schreiber, G., and Fersht, A.R. 1994. Protein–protein recognition: Crystal structural analysis of a barnase–barstar complex at 2.0-A resolution. Biochemistry 33 8878–8889. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
