

Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes
- PMID: 11581301
- PMCID: PMC200951
- DOI: 10.1172/JCI12864
Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes
Abstract
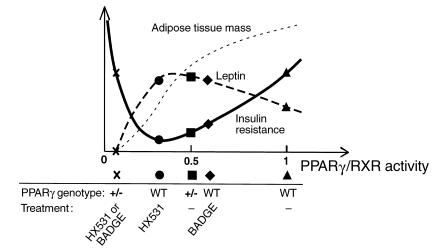
PPARgamma is a ligand-activated transcription factor and functions as a heterodimer with a retinoid X receptor (RXR). Supraphysiological activation of PPARgamma by thiazolidinediones can reduce insulin resistance and hyperglycemia in type 2 diabetes, but these drugs can also cause weight gain. Quite unexpectedly, a moderate reduction of PPARgamma activity observed in heterozygous PPARgamma-deficient mice or the Pro12Ala polymorphism in human PPARgamma, has been shown to prevent insulin resistance and obesity induced by a high-fat diet. In this study, we investigated whether functional antagonism toward PPARgamma/RXR could be used to treat obesity and type 2 diabetes. We show herein that an RXR antagonist and a PPARgamma antagonist decrease triglyceride (TG) content in white adipose tissue, skeletal muscle, and liver. These inhibitors potentiated leptin's effects and increased fatty acid combustion and energy dissipation, thereby ameliorating HF diet-induced obesity and insulin resistance. Paradoxically, treatment of heterozygous PPARgamma-deficient mice with an RXR antagonist or a PPARgamma antagonist depletes white adipose tissue and markedly decreases leptin levels and energy dissipation, which increases TG content in skeletal muscle and the liver, thereby leading to the re-emergence of insulin resistance. Our data suggested that appropriate functional antagonism of PPARgamma/RXR may be a logical approach to protection against obesity and related diseases such as type 2 diabetes.
Figures
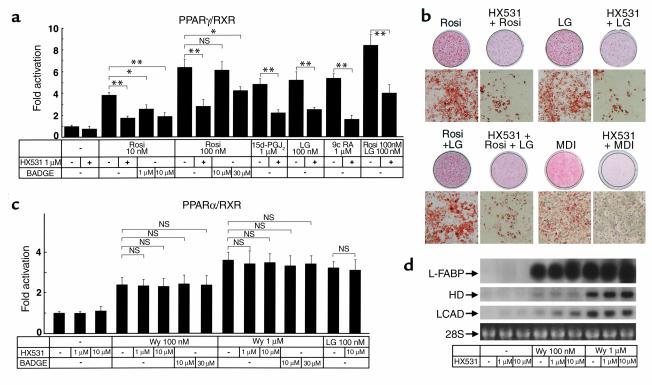
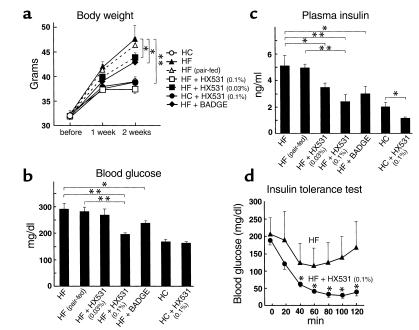
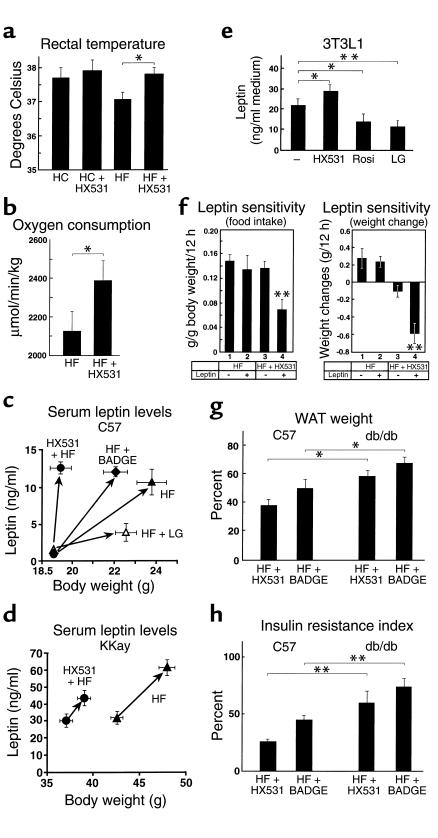
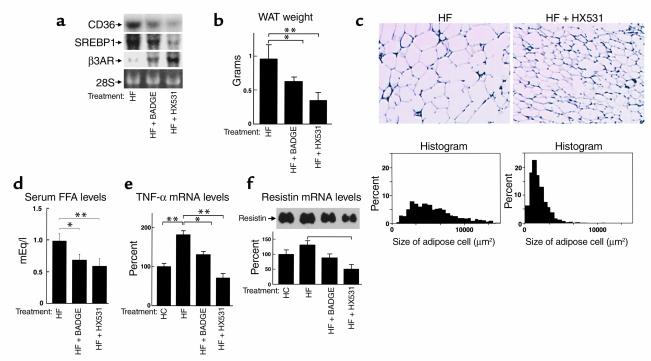
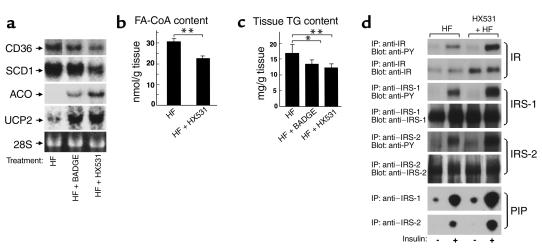
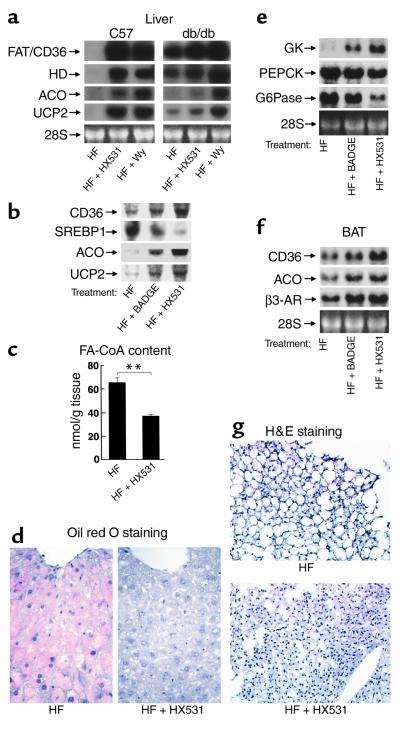
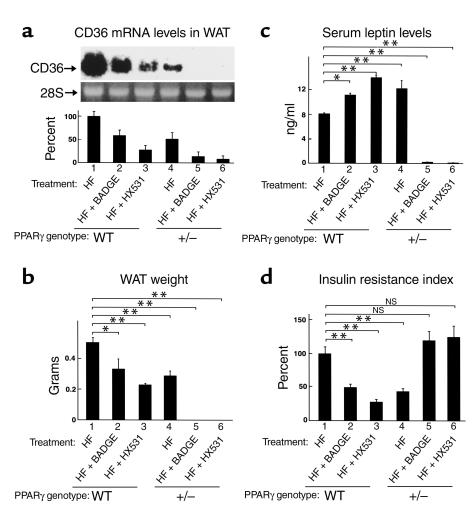
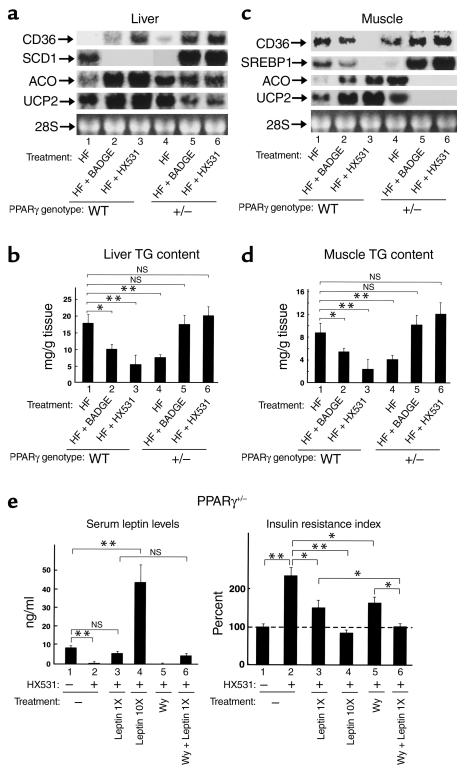
References
-
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. - PubMed
-
- Lowell BB. PPARgamma: an essential regulator of adipogenesis and modulator of fat cell function. Cell. 1999;99:239–242. - PubMed
-
- Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. - PubMed
-
- Gonzalez FJ. Recent update on the PPAR alpha-null mouse. Biochimie. 1997;79:139–144. - PubMed
-
- Auwerx J. PPARgamma, the ultimate thrifty gene. Diabetologia. 1999;42:1033–1049. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
