

Restoration of nucleotide excision repair in a helicase-deficient XPD mutant from intragenic suppression by a trichothiodystrophy mutation
- PMID: 11585917
- PMCID: PMC99909
- DOI: 10.1128/MCB.21.21.7355-7365.2001
Restoration of nucleotide excision repair in a helicase-deficient XPD mutant from intragenic suppression by a trichothiodystrophy mutation
Abstract
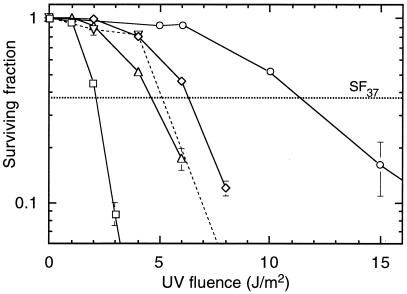
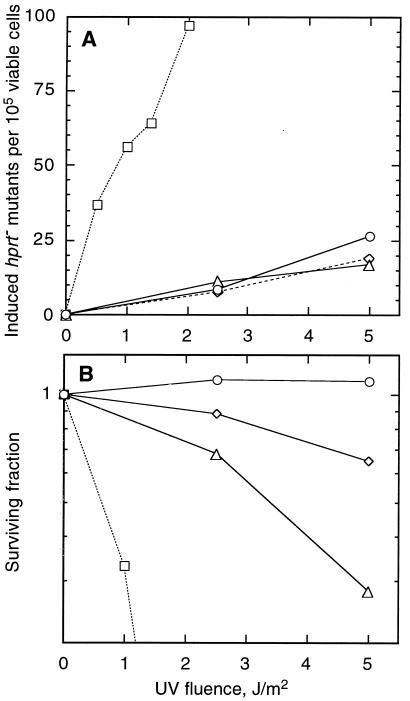
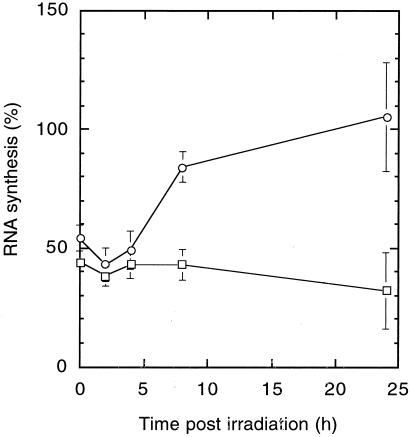
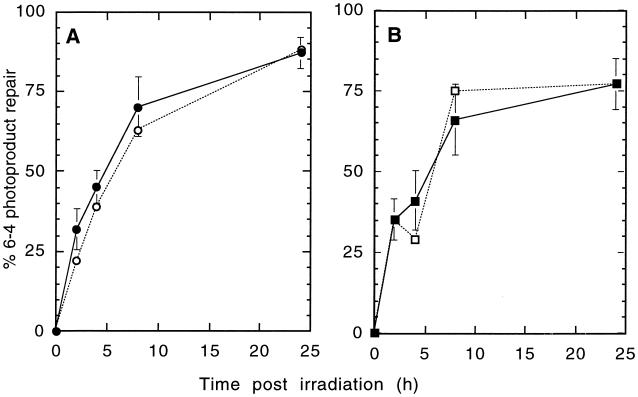
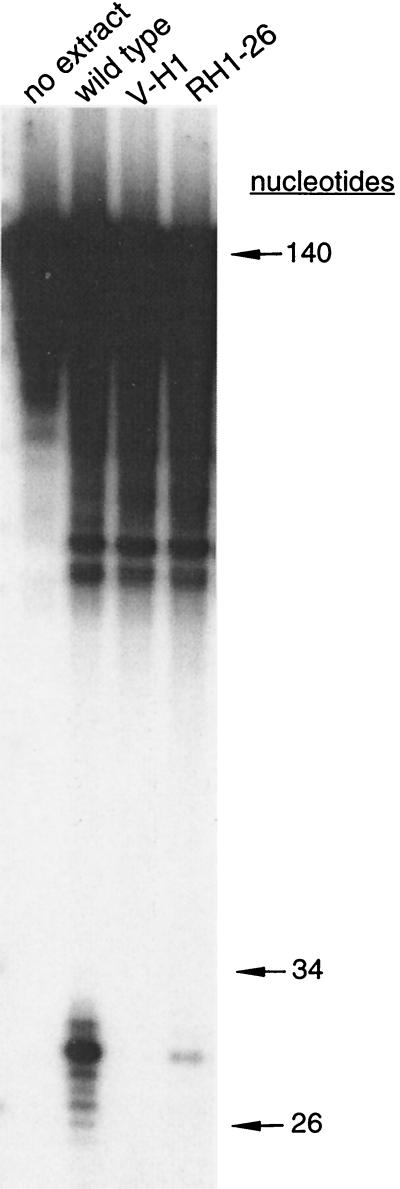
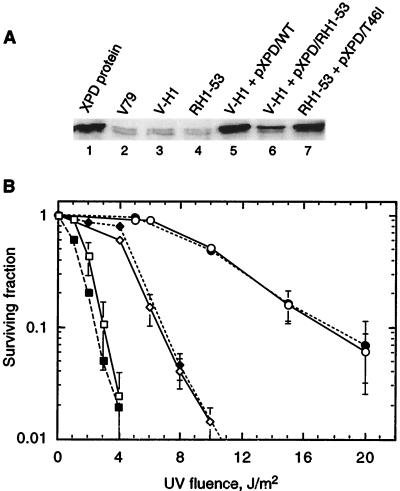
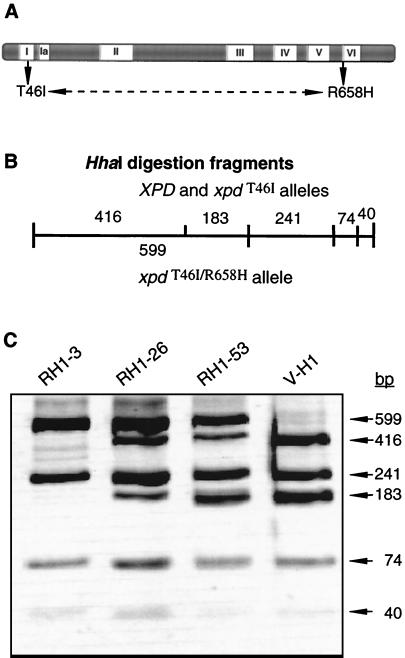
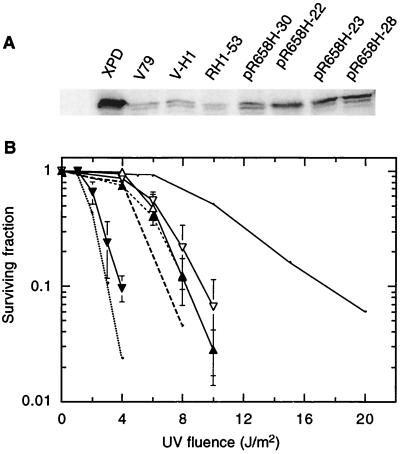
The UV-sensitive V-H1 cell line has a T46I substitution mutation in the Walker A box in both alleles of XPD and lacks DNA helicase activity. We characterized three partial revertants that curiously display intermediate UV cytotoxicity (2- to 2.5-fold) but normal levels of UV-induced hprt mutations. In revertant RH1-26, the efficient removal of pyrimidine (6-4) pyrimidone photoproducts from both strands of hprt suggests that global-genomic nucleotide excision repair is normal, but the pattern of cyclobutane pyrimidine dimer removal suggests that transcription-coupled repair (TCR) is impaired. To explain the intermediate UV survival and lack of RNA synthesis recovery in RH1-26 after 10 J of UV/m(2), we propose a defect in repair-transcription coupling, i.e., the inability of the cells to resume or reinitiate transcription after the first TCR event within a transcript. All three revertants carry an R658H suppressor mutation, in one allele of revertants RH1-26 and RH1-53 and in both alleles of revertant RH1-3. Remarkably, the R658H mutation produces the clinical phenotype of trichothiodystrophy (TTD) in several patients who display intermediate UV sensitivity. The XPD(R658H) TTD protein, like XPD(T46I/R658H), is codominant when overexpressed in V-H1 cells and partially complements their UV sensitivity. Thus, the suppressing R658H substitution must restore helicase activity to the inactive XPD(T46I) protein. Based on current knowledge of helicase structure, the intragenic reversion mutation may partially compensate for the T46I mutation by perturbing the XPD structure in a way that counteracts the effect of this mutation. These findings have implications for understanding the differences between xeroderma pigmentosum and TTD and illustrate the value of suppressor genetics for studying helicase structure-function relationships.
Figures
Similar articles
-
Mouse model for the DNA repair/basal transcription disorder trichothiodystrophy reveals cancer predisposition.Cancer Res. 1999 Jul 15;59(14):3489-94. Cancer Res. 1999. PMID: 10416615
-
Effects of XPD mutations on ultraviolet-induced apoptosis in relation to skin cancer-proneness in repair-deficient syndromes.J Invest Dermatol. 2001 Nov;117(5):1162-70. doi: 10.1046/j.0022-202x.2001.01533.x. J Invest Dermatol. 2001. PMID: 11710928
-
Codominance associated with overexpression of certain XPD mutations.Mutat Res. 2001 Mar 7;485(2):153-68. doi: 10.1016/s0921-8777(00)00077-x. Mutat Res. 2001. PMID: 11182546
-
Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes.J Am Acad Dermatol. 2001 Jun;44(6):891-920; quiz 921-4. doi: 10.1067/mjd.2001.114294. J Am Acad Dermatol. 2001. PMID: 11369901 Review.
-
Xeroderma pigmentosum and molecular cloning of DNA repair genes.Anticancer Res. 1996 Mar-Apr;16(2):693-708. Anticancer Res. 1996. PMID: 8687116 Review.
Cited by
-
A Simple, Rapid, and Quantitative Assay to Measure Repair of DNA-protein Crosslinks on Plasmids Transfected into Mammalian Cells.J Vis Exp. 2018 Mar 5;(133):57413. doi: 10.3791/57413. J Vis Exp. 2018. PMID: 29553515 Free PMC article.
-
A quantitative PCR-based assay reveals that nucleotide excision repair plays a predominant role in the removal of DNA-protein crosslinks from plasmids transfected into mammalian cells.DNA Repair (Amst). 2018 Feb;62:18-27. doi: 10.1016/j.dnarep.2018.01.004. Epub 2018 Jan 9. DNA Repair (Amst). 2018. PMID: 29413806 Free PMC article.
-
Somatic genetic rescue in Mendelian haematopoietic diseases.Nat Rev Genet. 2019 Oct;20(10):582-598. doi: 10.1038/s41576-019-0139-x. Epub 2019 Jun 11. Nat Rev Genet. 2019. PMID: 31186537 Review.
References
-
- Araujo S J, Wood R D. Protein complexes in nucleotide excision repair. Mutat Res. 1999;435:23–33. - PubMed
-
- Balajee A S, DeSantis L P, Brosh Jr R M, Selzer R, Bohr V A. Role of the ATPase domain of the Cockayne syndrome group B protein in UV induced apoptosis. Oncogene. 2000;19:477–489. - PubMed
-
- Berneburg M, Lehmann A R. Xeroderma pigmentosum and related disorders: defects in DNA repair and transcription. Adv Genet. 2001;43:71–102. - PubMed
-
- Bootsma D, Hoeijmakers J H. The molecular basis of nucleotide excision repair syndromes. Mutat Res. 1994;307:15–23. - PubMed
-
- Bootsma D, Kraemer K H, Cleaver J E, Hoeijmakers J H J. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler K, editors. The genetic basis of human cancer. New York, N.Y: McGraw-Hill Book Co.; 1998. pp. 245–274.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous