

Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome
- PMID: 11591643
- PMCID: PMC311155
- DOI: 10.1101/gr.188201
Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome
Abstract
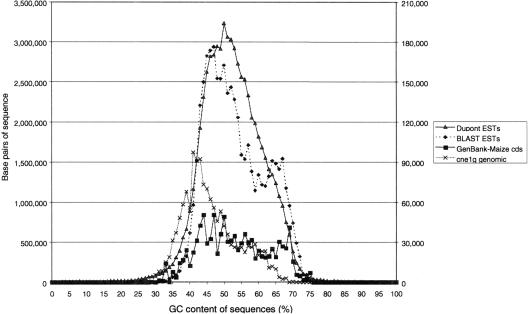
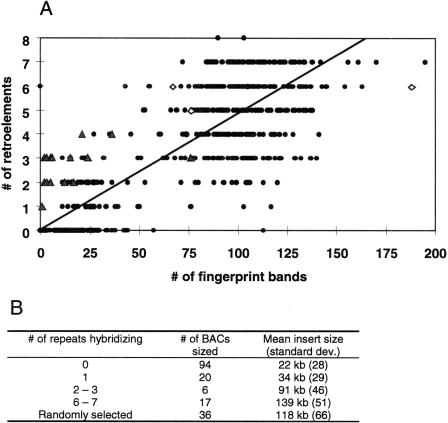
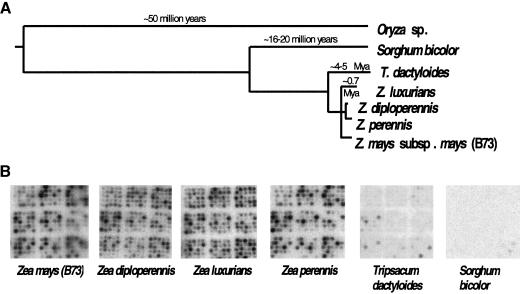
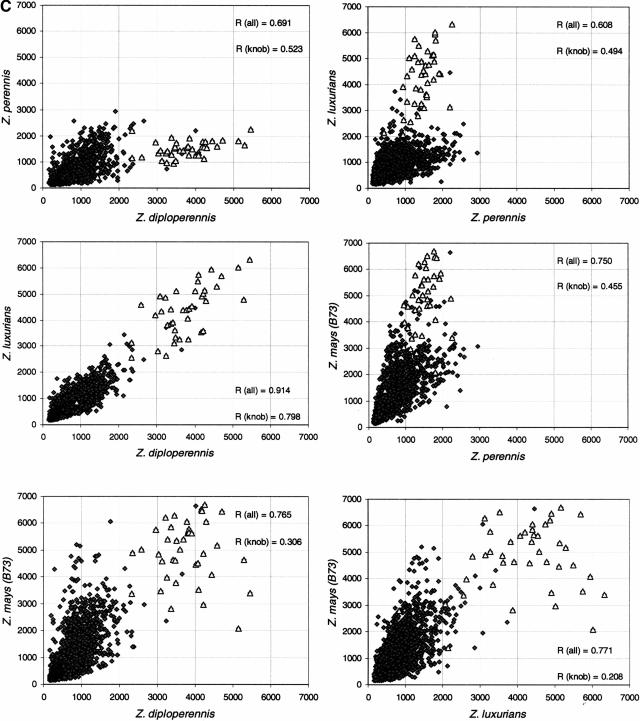
Long terminal repeat (LTR) retrotransposons have been shown to make up much of the maize genome. Although these elements are known to be prevalent in plant genomes of a middle-to-large size, little information is available on the relative proportions composed by specific families of elements in a single genome. We sequenced a library of randomly sheared genomic DNA from maize to characterize this genome. BLAST analysis of these sequences demonstrated that the maize genome is composed of diverse sequences that represent numerous families of retrotransposons. The largest families contain the previously described elements Huck, Ji, and Opie. Approximately 5% of the sequences are predicted to encode proteins. The genomic abundance of 16 families of elements was estimated by hybridization to an array of 10,752 maize bacterial artificial chromosome (BAC) clones. Comparisons of the number of elements present on individual BACs indicated that retrotransposons are in general randomly distributed across the maize genome. A second library was constructed that was selected to contain sequences hypomethylated in the maize genome. Sequence analysis of this library indicated that retroelements abundant in the genome are poorly represented in hypomethylated regions. Fifty-six retroelement sequences corresponding to the integrase and reverse transcriptase domains were isolated from approximately 407,000 maize expressed sequence tags (ESTs). Phylogenetic analysis of these and the genomic retroelement sequences indicated that elements most abundant in the genome are less abundant at the transcript level than are more rare retrotransposons. Additional phylogenies also demonstrated that rice and maize retrotransposon families are frequently more closely related to each other than to families within the same species. An analysis of the GC content of the maize genomic library and that of maize ESTs did not support recently published data that the gene space in maize is found within a narrow GC range, but does indicate that genic sequences have a higher GC content than intergenic sequences (52% vs. 47% GC).
Figures
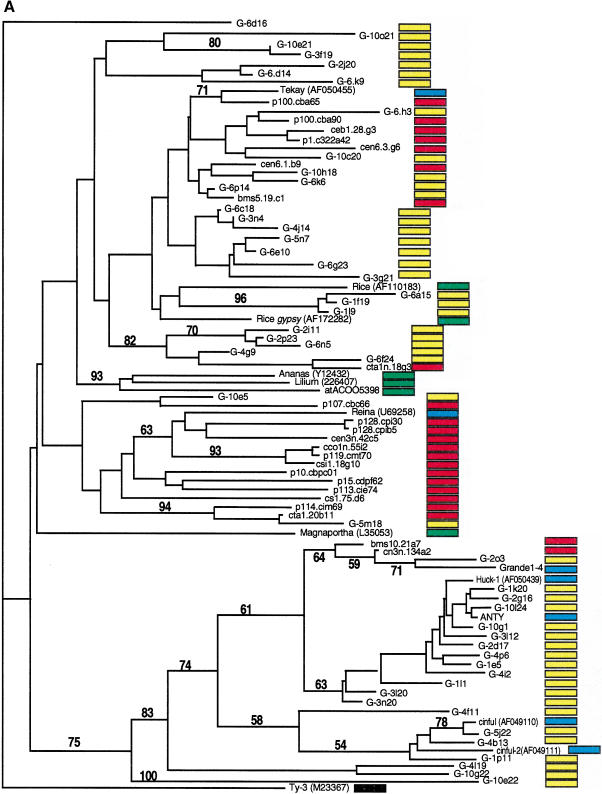
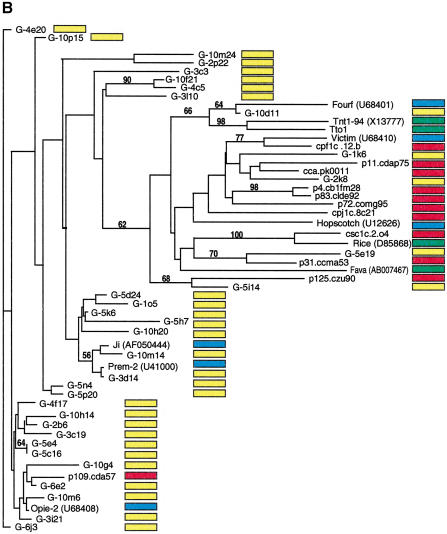
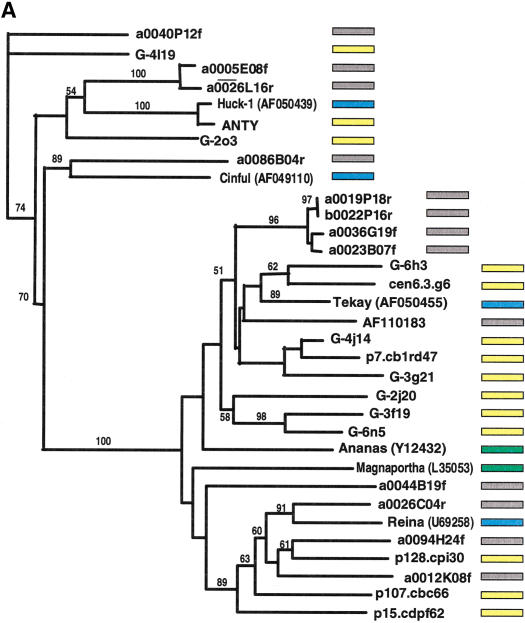
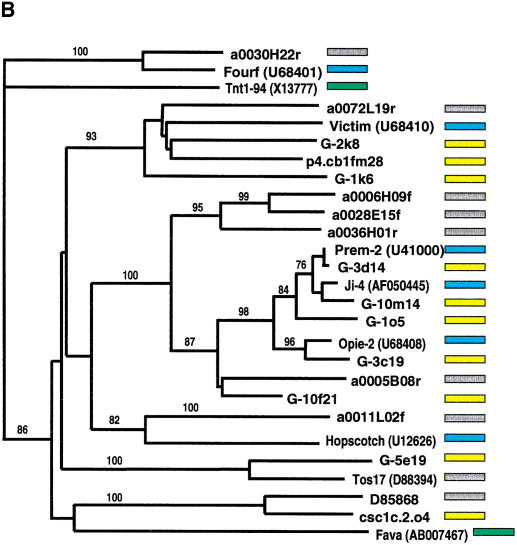
References
-
- Arumuganathan K, Earle ED. Nuclear DNA content of some important plant species. Plant Mol Biol Rep. 1991;9:208–218.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous