

Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features
- PMID: 11592033
- PMCID: PMC1235530
- DOI: 10.1086/324343
Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features
Abstract
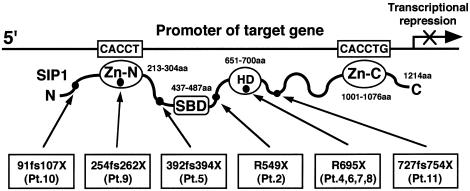
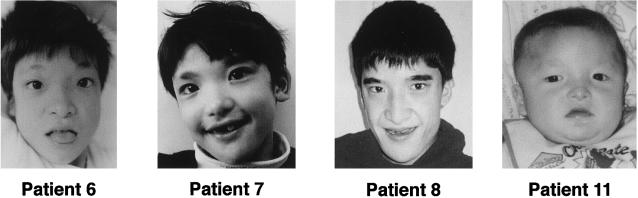
Mutations in ZFHX1B, encoding Smad-interacting protein 1 (SIP1), have been recently reported to cause a form of Hirschsprung disease (HSCR). Patients with ZFHX1B deficiency typically show mental retardation, delayed motor development, epilepsy, microcephaly, distinct facial features, and/or congenital heart disease, in addition to the cardinal form of HSCR. To investigate the breadth of clinical variation, we studied DNA samples from six patients with clinical profiles quite similar to those described elsewhere for ZFHX1B deficiency, except that they did not have HSCR. The results showed the previously reported R695X mutation to be present in three cases, with three novel mutations-a 2-bp insertion (760insCA resulting in 254fs262X), a single-base deletion (270delG resulting in 91fs107X), and a 2-bp deletion (2178delTT resulting in 727fs754X)-newly identified in the other three. All mutations occurred in one allele and were de novo events. These results demonstrate that ZFHX1B deficiency is an autosomal dominant complex developmental disorder and that individuals with functional null mutations present with mental retardation, delayed motor development, epilepsy, and a wide spectrum of clinically heterogeneous features suggestive of neurocristopathies at the cephalic, cardiac, and vagal levels.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for ZFHX1B mRNA [accession number AB 056507] and Zfx1b [Mus musculus] [accession number AF 033116])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HSCR [MIM 142623], HSCR with microcephaly, mental retardation, and distinct facial features [MIM 235730], and ZFHX1B, encoding SIP1 [MIM 605802])
References
-
- Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A (1996) Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet 14:341–344 - PubMed
-
- Cacheux V, Dastot-Le Moal F, Kääriäinen H, Bondurand N, Rintala R, Boissier B, Wilson M, Mowat D, Goossens M (2001) Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet 10:1503–1510 - PubMed
-
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278 - PubMed
-
- Cooper DN, Youssoufian H (1988) The CpG dinucleotide and human genetic disease. Hum Genet 78:151–155 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
