

Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury
- PMID: 11606641
- PMCID: PMC6762822
- DOI: 10.1523/JNEUROSCI.21-21-08523.2001
Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury
Abstract
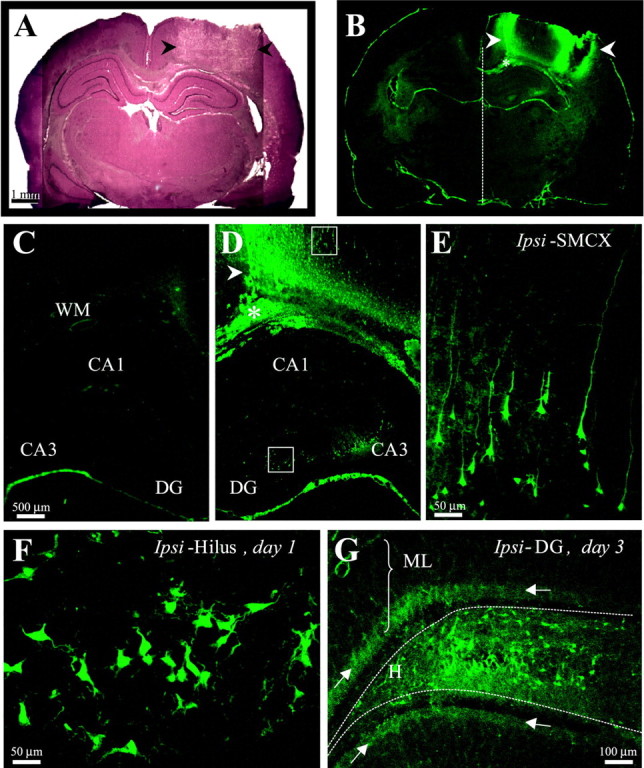
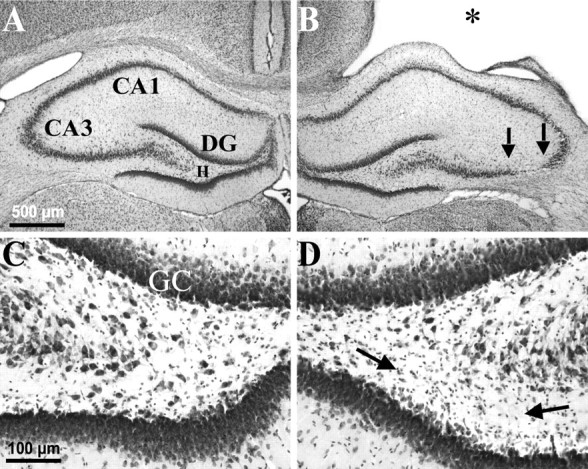
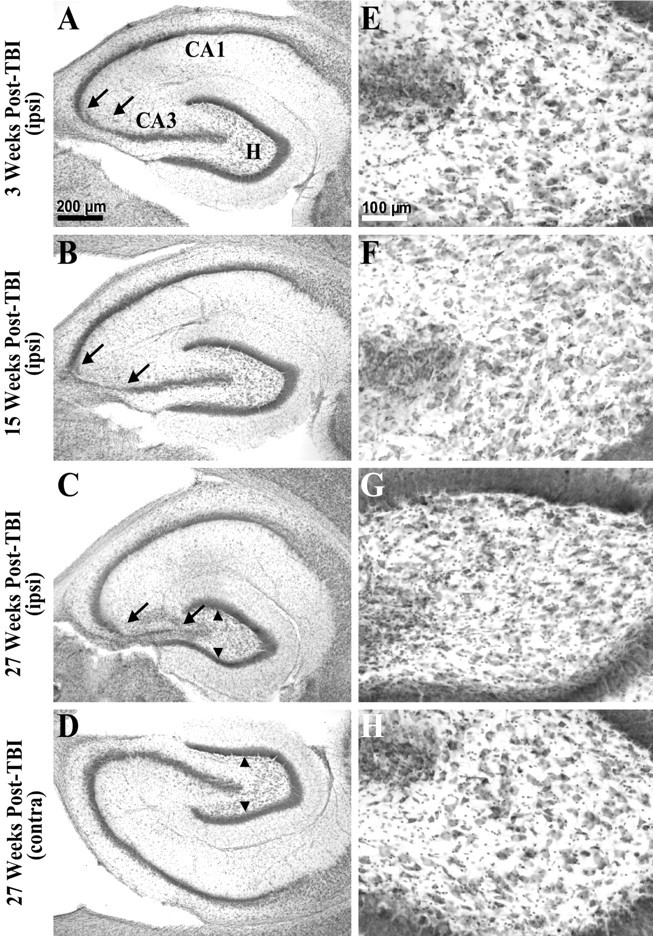
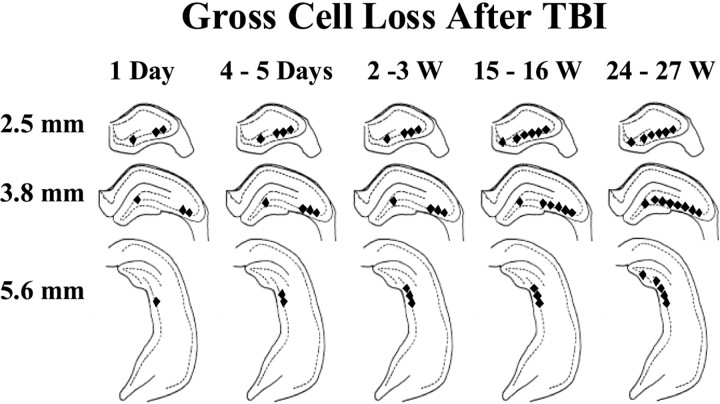
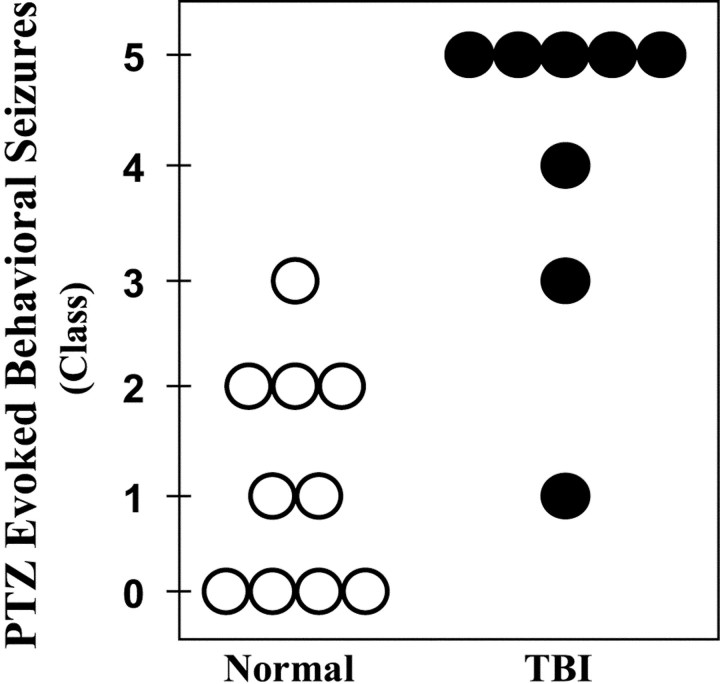
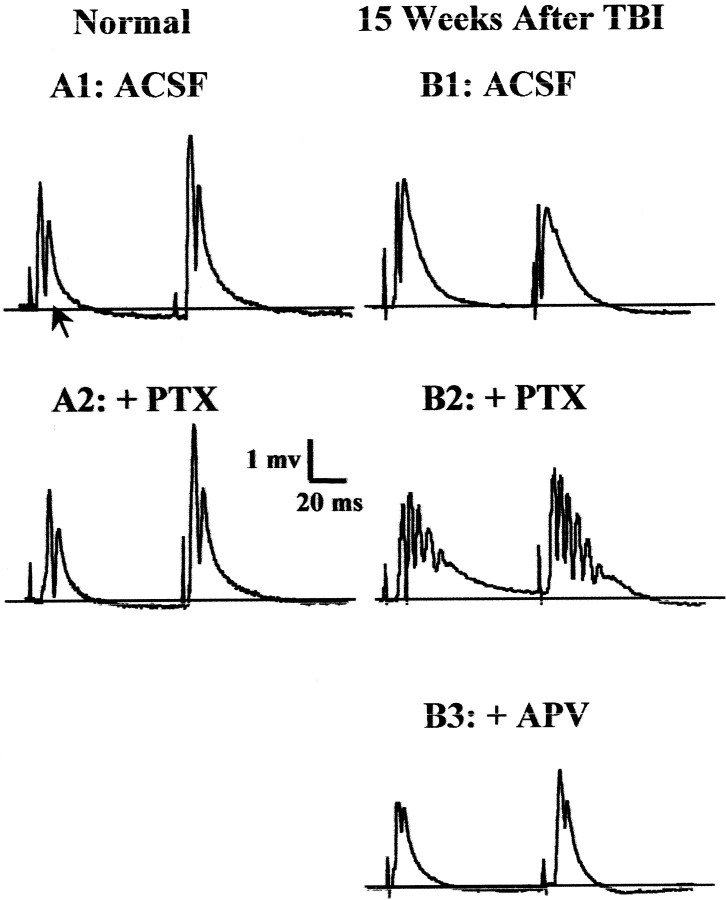
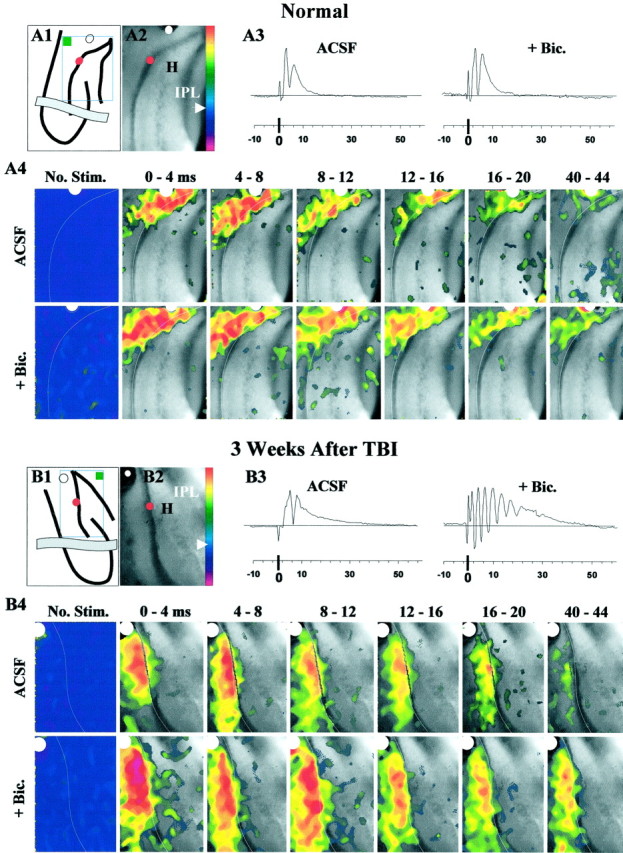
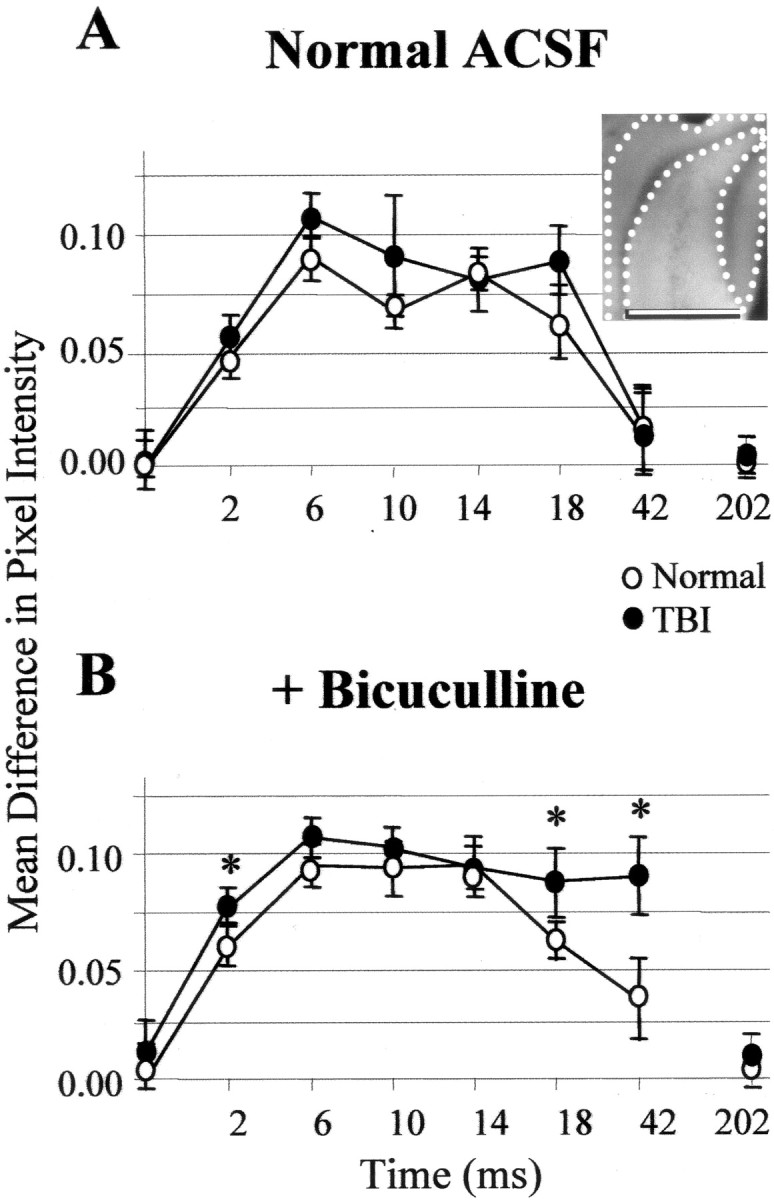
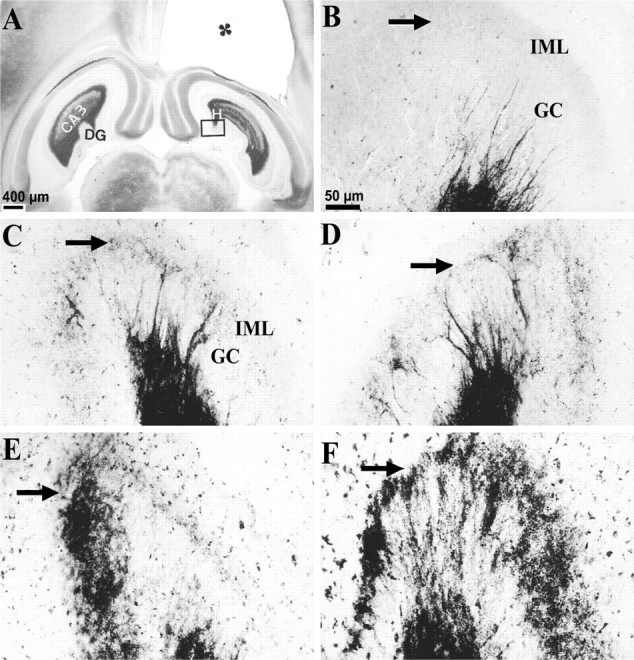
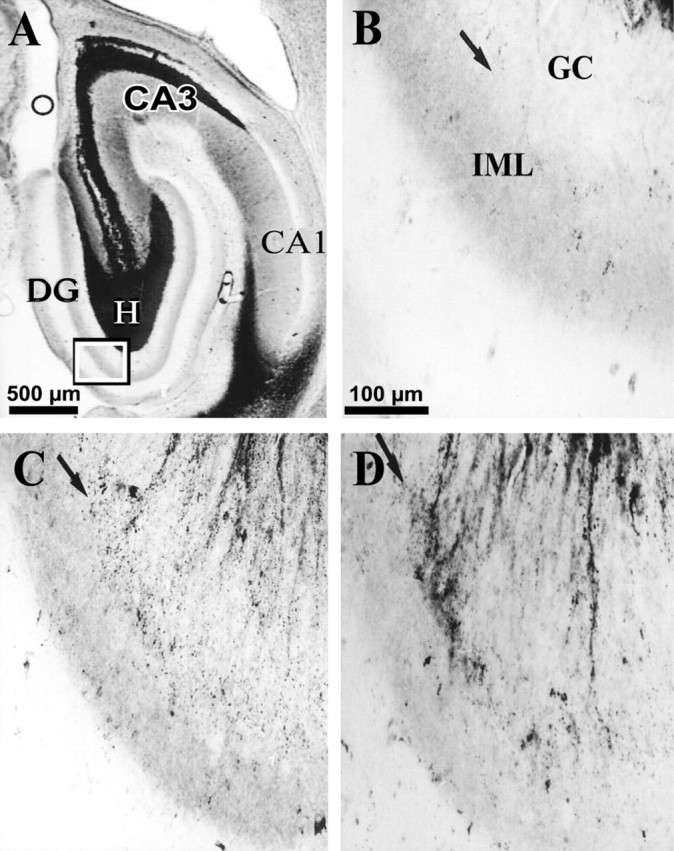
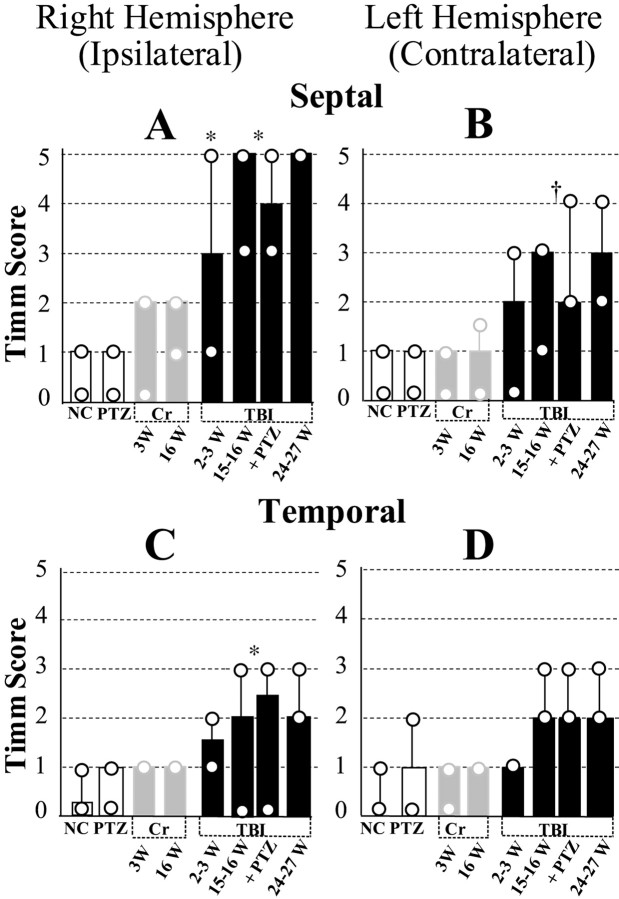
Epilepsy is a common outcome of traumatic brain injury (TBI), but the mechanisms of posttraumatic epileptogenesis are poorly understood. One clue is the occurrence of selective hippocampal cell death after fluid-percussion TBI in rats, consistent with the reported reduction of hippocampal volume bilaterally in humans after TBI and resembling hippocampal sclerosis, a hallmark of temporal-lobe epilepsy. Other features of temporal-lobe epilepsy, such as long-term seizure susceptibility, persistent hyperexcitability in the dentate gyrus (DG), and mossy fiber synaptic reorganization, however, have not been examined after TBI. To determine whether TBI induces these changes, we used a well studied model of TBI by weight drop on somatosensory cortex in adult rats. First, we confirmed an early and selective cell loss in the hilus of the DG and area CA3 of hippocampus, ipsilateral to the impact. Second, we found persistently enhanced susceptibility to pentylenetetrazole-induced convulsions 15 weeks after TBI. Third, by applying GABA(A) antagonists during field-potential and optical recordings in hippocampal slices 3 and 15 weeks after TBI, we unmasked a persistent, abnormal APV-sensitive hyperexcitability that was bilateral and localized to the granule cell and molecular layers of the DG. Finally, using Timm histochemistry, we detected progressive sprouting of mossy fibers into the inner molecular layers of the DG bilaterally 2-27 weeks after TBI. These findings are consistent with the development of posttraumatic epilepsy in an animal model of impact head injury, showing a striking similarity to the enduring behavioral, functional, and structural alterations associated with temporal-lobe epilepsy.
Figures
References
-
- Aamodt SM, Constantine-Paton AU. The role of neural activity in synaptic development and its implications for adult brain function. Adv Neurol. 1999;79:133–144. - PubMed
-
- Annegers JF, Rocca WA, Hauser WA. Causes of epilepsy: contributions of the Rochester epidemiology project. Mayo Clin Proc. 1996;71:570–575. - PubMed
-
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. - PubMed
-
- Asikainen I, Kaste M, Sarna S. Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia. 1999;40:584–589. - PubMed
-
- Babb TL. Axonal growth and neosynaptogenesis in human and experimental hippocampal epilepsy. Adv Neurol. 1997;72:45–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous