

Phosphorylation of serines 635 and 645 of human Rad17 is cell cycle regulated and is required for G(1)/S checkpoint activation in response to DNA damage
- PMID: 11687627
- PMCID: PMC60831
- DOI: 10.1073/pnas.231364598
Phosphorylation of serines 635 and 645 of human Rad17 is cell cycle regulated and is required for G(1)/S checkpoint activation in response to DNA damage
Abstract
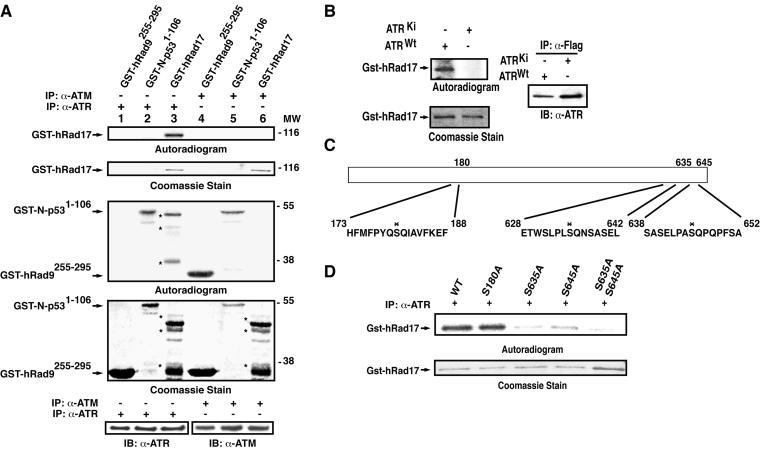
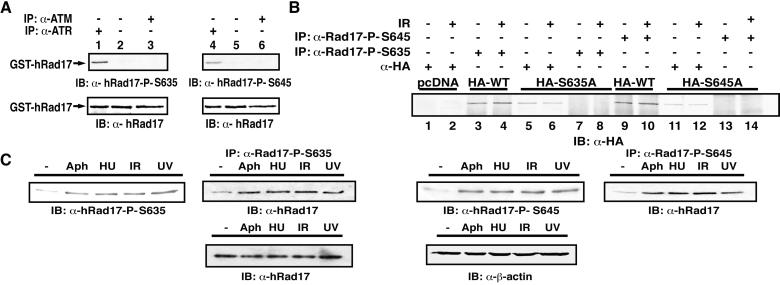
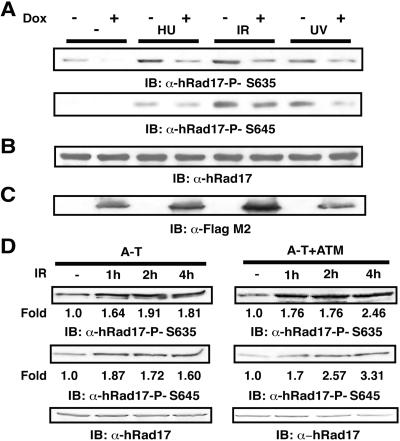
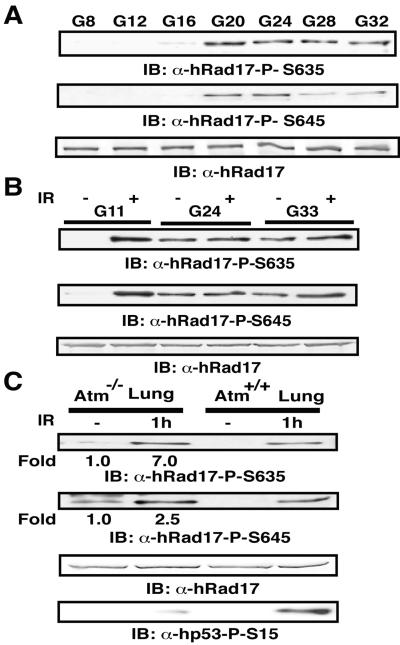
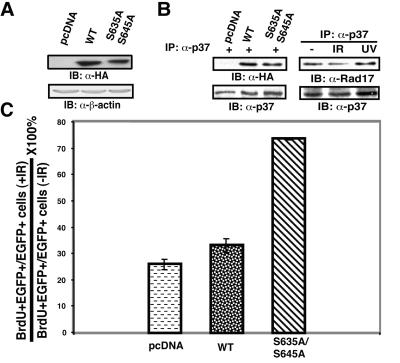
ATR [ataxia-telangiectasia-mutated (ATM)- and Rad3-related] is a protein kinase required for both DNA damage-induced cell cycle checkpoint responses and the DNA replication checkpoint that prevents mitosis before the completion of DNA synthesis. Although ATM and ATR kinases share many substrates, the different phenotypes of ATM- and ATR-deficient mice indicate that these kinases are not functionally redundant. Here we demonstrate that ATR but not ATM phosphorylates the human Rad17 (hRad17) checkpoint protein on Ser(635) and Ser(645) in vitro. In undamaged synchronized human cells, these two sites were phosphorylated in late G(1), S, and G(2)/M, but not in early-mid G(1). Treatment of cells with genotoxic stress induced phosphorylation of hRad17 in cells in early-mid G(1). Expression of kinase-inactive ATR resulted in reduced phosphorylation of these residues, but these same serine residues were phosphorylated in ionizing radiation (IR)-treated ATM-deficient human cell lines. IR-induced phosphorylation of hRad17 was also observed in ATM-deficient tissues, but induction of Ser(645) was not optimal. Expression of a hRad17 mutant, with both serine residues changed to alanine, abolished IR-induced activation of the G(1)/S checkpoint in MCF-7 cells. These results suggest ATR and hRad17 are essential components of a DNA damage response pathway in mammalian cells.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
