

Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun
- PMID: 11689607
- PMCID: PMC114712
- DOI: 10.1128/JVI.75.23.11275-11283.2001
Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun
Abstract
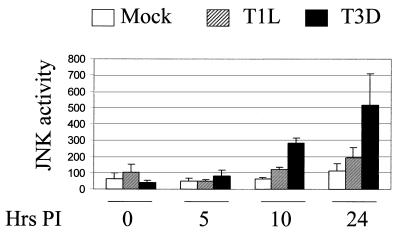
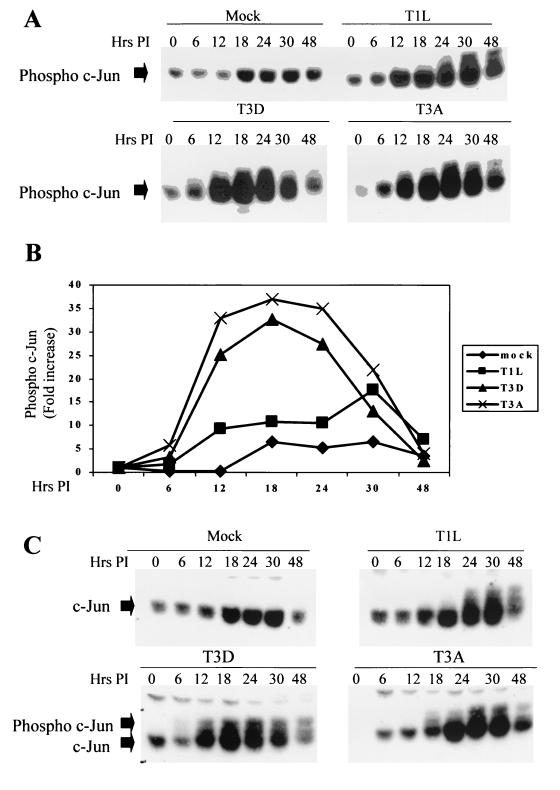
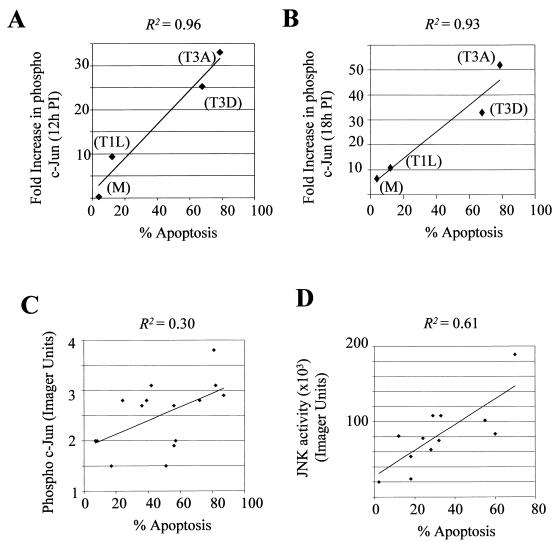
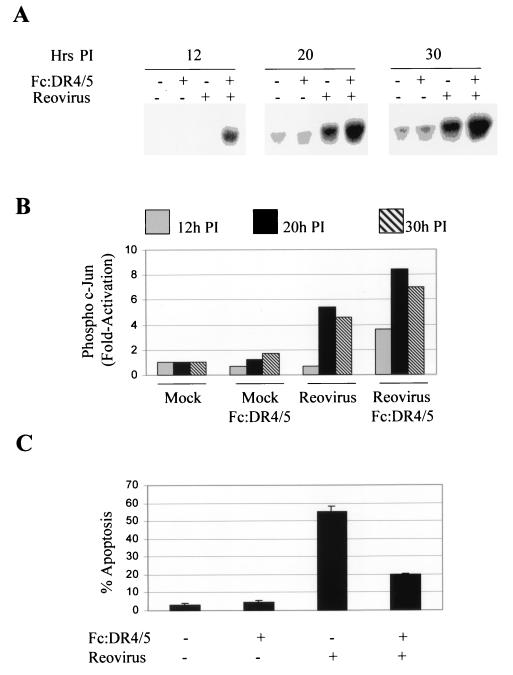
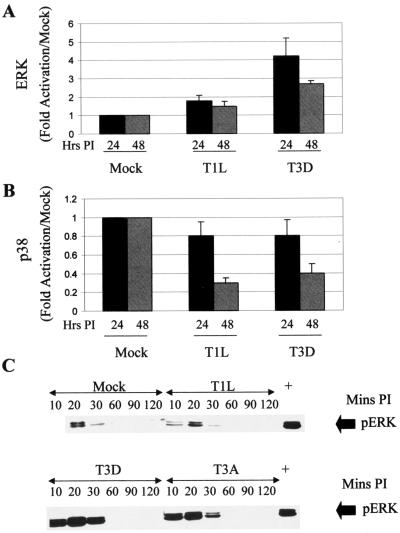
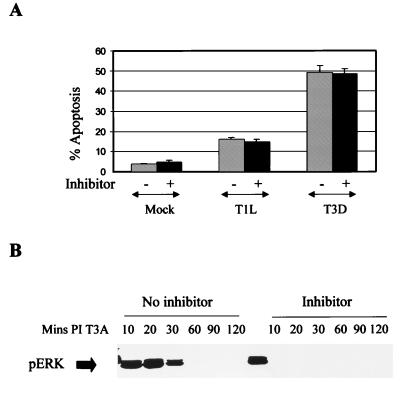
Viral infection often perturbs host cell signaling pathways including those involving mitogen-activated protein kinases (MAPKs). We now show that reovirus infection results in the selective activation of c-Jun N-terminal kinase (JNK). Reovirus-induced JNK activation is associated with an increase in the phosphorylation of the JNK-dependent transcription factor c-Jun. Reovirus serotype 3 prototype strains Abney (T3A) and Dearing (T3D) induce significantly more JNK activation and c-Jun phosphorylation than does the serotype 1 prototypic strain Lang (T1L). T3D and T3A also induce more apoptosis in infected cells than T1L, and there was a significant correlation between the ability of these viruses to phosphorylate c-Jun and induce apoptosis. However, reovirus-induced apoptosis, but not reovirus-induced c-Jun phosphorylation, is inhibited by blocking TRAIL/receptor binding, suggesting that apoptosis and c-Jun phosphorylation involve parallel rather than identical pathways. Strain-specific differences in JNK activation are determined by the reovirus S1 and M2 gene segments, which encode viral outer capsid proteins (sigma1 and mu1c) involved in receptor binding and host cell membrane penetration. These same gene segments also determine differences in the capacity of reovirus strains to induce apoptosis, and again a significant correlation between the capacity of T1L x T3D reassortant reoviruses to both activate JNK and phosphorylate c-Jun and to induce apoptosis was shown. The extracellular signal-related kinase (ERK) is also activated in a strain-specific manner following reovirus infection. Unlike JNK activation, ERK activation could not be mapped to specific reovirus gene segments, suggesting that ERK activation and JNK activation are triggered by different events during virus-host cell interaction.
Figures
Similar articles
-
Reovirus receptors, cell entry, and proapoptotic signaling.Adv Exp Med Biol. 2013;790:42-71. doi: 10.1007/978-1-4614-7651-1_3. Adv Exp Med Biol. 2013. PMID: 23884585 Free PMC article. Review.
-
Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes.J Virol. 1996 Nov;70(11):7984-91. doi: 10.1128/JVI.70.11.7984-7991.1996. J Virol. 1996. PMID: 8892922 Free PMC article.
-
Reovirus-induced G(2)/M cell cycle arrest requires sigma1s and occurs in the absence of apoptosis.J Virol. 2000 Oct;74(20):9562-70. doi: 10.1128/jvi.74.20.9562-9570.2000. J Virol. 2000. PMID: 11000227 Free PMC article.
-
Reovirus mu2 protein determines strain-specific differences in the rate of viral inclusion formation in L929 cells.Virology. 2000 Jun 20;272(1):16-26. doi: 10.1006/viro.2000.0362. Virology. 2000. PMID: 10873745
-
Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation.Viral Immunol. 2005;18(1):89-115. doi: 10.1089/vim.2005.18.89. Viral Immunol. 2005. PMID: 15802955 Free PMC article. Review.
Cited by
-
Reovirus receptors and pathogenesis.J Virol. 2003 Sep;77(17):9109-15. doi: 10.1128/jvi.77.17.9109-9115.2003. J Virol. 2003. PMID: 12915527 Free PMC article. Review. No abstract available.
-
Enter the kill zone: initiation of death signaling during virus entry.Virology. 2011 Mar 15;411(2):316-24. doi: 10.1016/j.virol.2010.12.043. Epub 2011 Jan 22. Virology. 2011. PMID: 21262519 Free PMC article. Review.
-
Activation of c-jun N-terminal kinase upon influenza A virus (IAV) infection is independent of pathogen-related receptors but dependent on amino acid sequence variations of IAV NS1.J Virol. 2014 Aug;88(16):8843-52. doi: 10.1128/JVI.00424-14. Epub 2014 May 28. J Virol. 2014. PMID: 24872593 Free PMC article.
-
Sigesbeckia orientalis L. Derived Active Fraction Ameliorates Perioperative Neurocognitive Disorders Through Alleviating Hippocampal Neuroinflammation.Front Pharmacol. 2022 Mar 17;13:846631. doi: 10.3389/fphar.2022.846631. eCollection 2022. Front Pharmacol. 2022. PMID: 35370714 Free PMC article.
-
Reovirus receptors, cell entry, and proapoptotic signaling.Adv Exp Med Biol. 2013;790:42-71. doi: 10.1007/978-1-4614-7651-1_3. Adv Exp Med Biol. 2013. PMID: 23884585 Free PMC article. Review.
References
-
- Ashkenazi A, Dixit V M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. - PubMed
-
- Barton E S, Forrest J C, Connolly J L, Chappell J D, Liu Y, Schnell F J, Nusrat A, Parkos C A, Dermody T S. Junction Adhesion Molecule is a receptor for reovirus. Cell. 2001;104:441–451. - PubMed
-
- Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. - PubMed
-
- Boldin M P, Varfolomeev E E, Pancer Z, Mett I L, Camonis J H, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
