

Identification of epilepsy genes in human and mouse
- PMID: 11700294
- PMCID: PMC2765248
- DOI: 10.1146/annurev.genet.35.102401.091142
Identification of epilepsy genes in human and mouse
Abstract
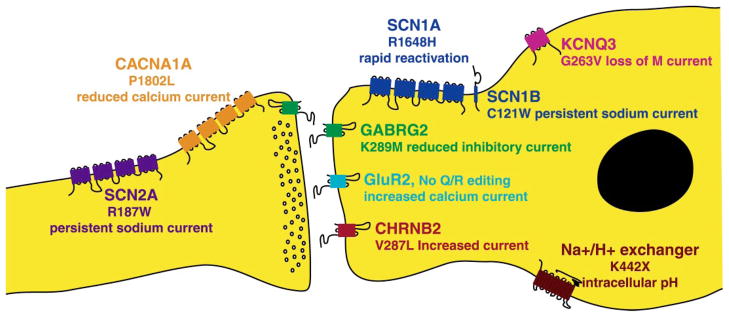
The development of molecular markers and genomic resources has facilitated the isolation of genes responsible for rare monogenic epilepsies in human and mouse. Many of the identified genes encode ion channels or other components of neuronal signaling. The electrophysiological properties of mutant alleles indicate that neuronal hyperexcitability is one cellular mechanism underlying seizures. Genetic heterogeneity and allelic variability are hallmarks of human epilepsy. For example, mutations in three different sodium channel genes can produce the same syndrome, GEFS+, while individuals with the same allele can experience different types of seizures. Haploinsufficiency for the sodium channel SCN1A has been demonstrated by the severe infantile epilepsy and cognitive deficits in heterozygotes for de novo null mutations. Large-scale patient screening is in progress to determine whether less severe alleles of the genes responsible for monogenic epilepsy may contribute to the common types of epilepsy in the human population. The development of pharmaceuticals directed towards specific epilepsy genotypes can be anticipated, and the introduction of patient mutations into the mouse genome will provide models for testing these targeted therapies.
Figures
References
-
- Abriel H, Wehrens XH, Benhorin J, Kerem B, Kass RS. Molecular pharmacology of the sodium channel mutation D1790G linked to the long-QT syndrome. Circulation. 2000;102:921–25. - PubMed
-
- Ashcroft F. Ion Channels and Disease. San Diego: Academic; 2000.
-
- Balaguero N, Barclay J, Mione M, Canti C, Brodbeck J, et al. Reduction in voltage-dependent calcium channel function in cerebellar purkinje cells of the mouse mutant ducky, which has a null mutation for the calcium channel accessory subunit α2δ2. Soc Neurosci Abstr. 2000;26:365.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
