

Can a place of origin of the main cystic fibrosis mutations be identified?
- PMID: 11713719
- PMCID: PMC384895
- DOI: 10.1086/338243
Can a place of origin of the main cystic fibrosis mutations be identified?
Abstract
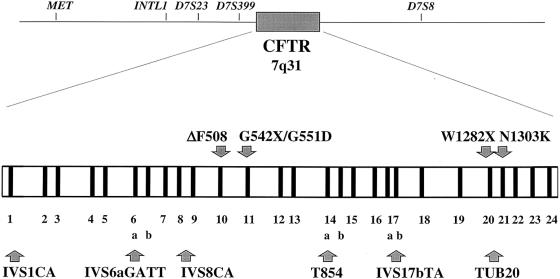
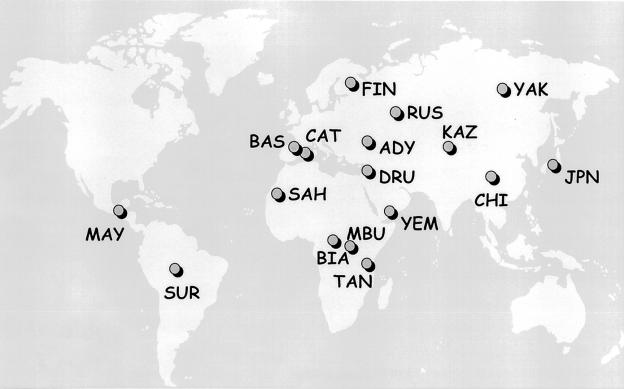
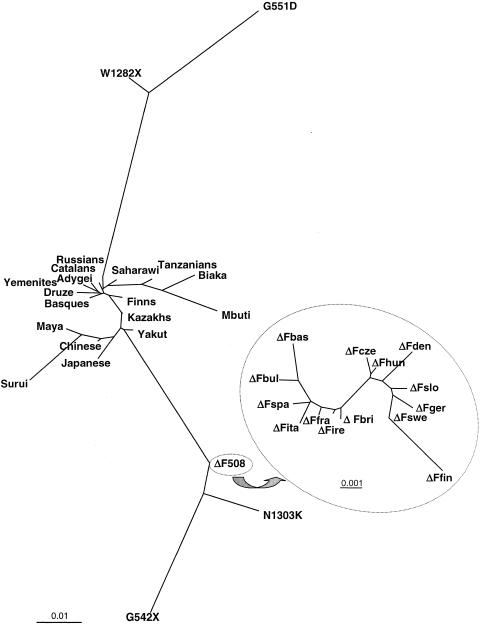
The genetic background of the mutations that most often cause cystic fibrosis (CF) is different from that of non-CF chromosomes in populations of European origin. It is not known whether these haplotype backgrounds could be found at high frequencies in populations in which CF is, at present, not common; such populations would be candidates for the place of origin of CF mutations. An analysis of haplotypes of CF transmembrane conductance regulator, together with their variation in specific CF chromosomes, in a worldwide survey of normal chromosomes shows (1) a very low frequency or absence of the most common CF haplotypes in all populations analyzed and (2) a strong genetic variability and divergence, among various populations, of the chromosomes that carry disease-causing mutations. The depth of the gene genealogy associated with disease-causing mutations may be greater than that of the evolutionary process that gave rise to present-day human populations. The concept of "population of origin" lacks either spatial or temporal meaning for mutations that are likely to have been present in Europeans before the ethnogenesis of present populations; subsequent population processes may have erased the traces of their geographic origin.
Figures
References
Electronic-Database Information
-
- Allele Frequency Database (ALFRED), http://info.med.yale.edu/genetics/kkidd (for further information on population samples)
-
- Cystic Fibrosis Mutation Data Base, http://www.genet.sickkids.on.ca/cftr
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CF [MIM 219700] and CFTR [MIM 602421])
References
-
- Bertranpetit J, Calafell F (1996) Genetic and geographical variability in cystic fibrosis: evolutionary considerations. In: Cardew G (ed) Variation in the human genome. Ciba Foundation Symposium 197. Wiley & Sons, Chichester, England, pp 97–118 - PubMed
-
- Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ
-
- Estivill X, Bancells C, Ramos C, Biomed CF Mutation Analysis Consortium (1997) Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. Hum Mutat 10:135–154 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
