

Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy
- PMID: 11714734
- PMCID: PMC209420
- DOI: 10.1172/JCI13350
Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy
Abstract
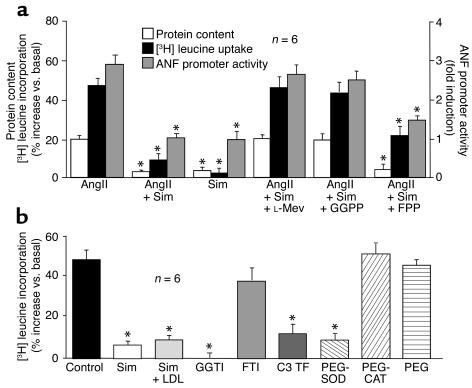
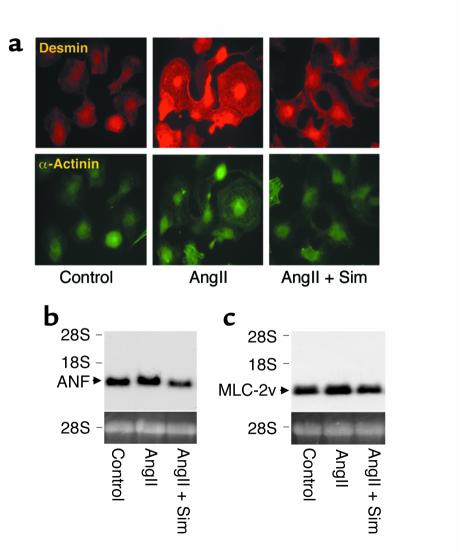
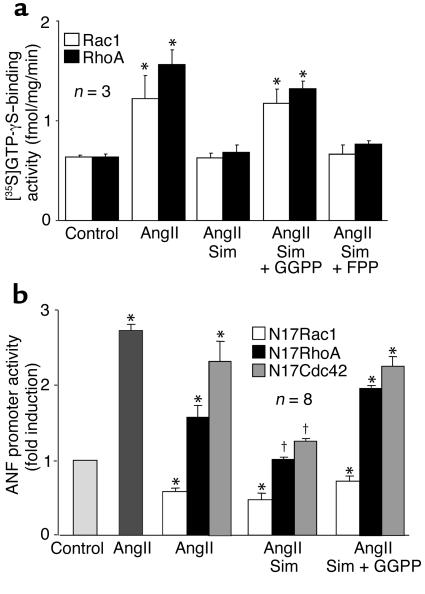
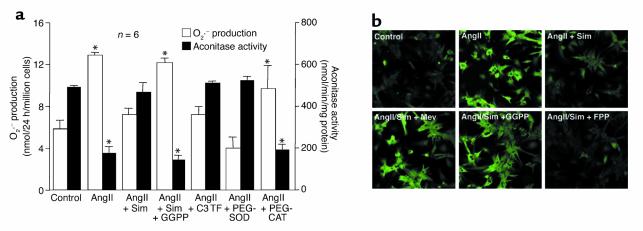
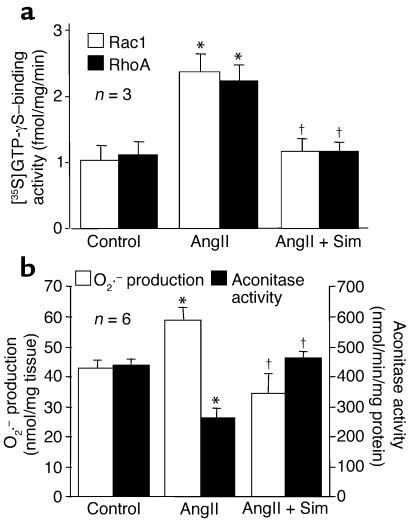
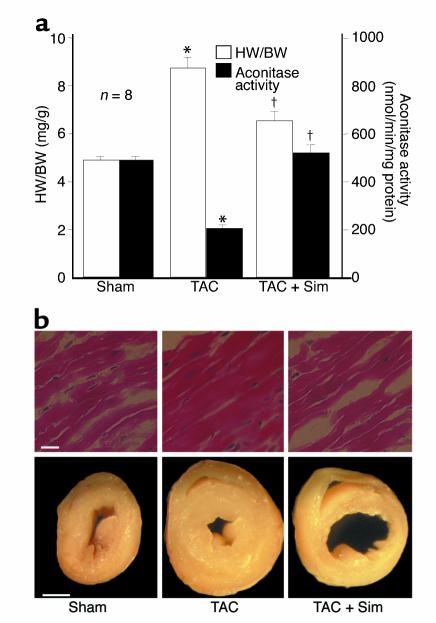
Cardiac hypertrophy is a major cause of morbidity and mortality worldwide. The hypertrophic process is mediated, in part, by small G proteins of the Rho family. We hypothesized that statins, inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase, inhibit cardiac hypertrophy by blocking Rho isoprenylation. We treated neonatal rat cardiac myocytes with angiotensin II (AngII) with and without simvastatin (Sim) and found that Sim decreased AngII-induced protein content, [3H] leucine uptake, and atrial natriuretic factor (ANF) promoter activity. These effects were associated with decreases in cell size, membrane Rho activity, superoxide anion (O2*-) production, and intracellular oxidation, and were reversed with L-mevalonate or geranylgeranylpyrophosphate, but not with farnesylpyrophosphate or cholesterol. Treatments with the Rho inhibitor C3 exotoxin and with cell-permeable superoxide dismutase also decreased AngII-induced O2*- production and myocyte hypertrophy. Overexpression of the dominant-negative Rho mutant N17Rac1 completely inhibited AngII-induced intracellular oxidation and ANF promoter activity, while N19RhoA partially inhibited it, and N17Cdc42 had no effect. Indeed, Sim inhibited cardiac hypertrophy and decreased myocardial Rac1 activity and O2*- production in rats treated with AngII infusion or subjected to transaortic constriction. These findings suggest that statins prevent the development of cardiac hypertrophy through an antioxidant mechanism involving inhibition of Rac1.
Figures

Similar articles
-
Impact of HMG CoA reductase inhibition on small GTPases in the heart.Cardiovasc Res. 2002 Mar;53(4):911-20. doi: 10.1016/s0008-6363(01)00540-5. Cardiovasc Res. 2002. PMID: 11922901
-
NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy.J Mol Cell Cardiol. 2003 Jul;35(7):851-9. doi: 10.1016/s0022-2828(03)00145-7. J Mol Cell Cardiol. 2003. PMID: 12818576
-
Association of RhoGDIalpha with Rac1 GTPase mediates free radical production during myocardial hypertrophy.Cardiovasc Res. 2006 Jul 15;71(2):342-51. doi: 10.1016/j.cardiores.2006.04.005. Epub 2006 Apr 19. Cardiovasc Res. 2006. PMID: 16698001
-
A novel pleiotropic effect of statins: prevention of cardiac hypertrophy by cholesterol-independent mechanisms.Ann Med. 2003;35(6):398-403. doi: 10.1080/07853890310001294. Ann Med. 2003. PMID: 14572163 Free PMC article. Review.
-
Statins and the myocardium.Semin Vasc Med. 2004 Nov;4(4):377-84. doi: 10.1055/s-2004-869594. Semin Vasc Med. 2004. PMID: 15861318 Free PMC article. Review.
Cited by
-
Atorvastatin reduces vascular endothelial growth factor (VEGF) expression in human non-small cell lung carcinomas (NSCLCs) via inhibition of reactive oxygen species (ROS) production.Mol Oncol. 2012 Feb;6(1):62-72. doi: 10.1016/j.molonc.2011.11.003. Epub 2011 Nov 22. Mol Oncol. 2012. PMID: 22153388 Free PMC article.
-
Pleiotropic effects of statins on acute kidney injury: involvement of Krüppel-like factor 4.Clin Exp Nephrol. 2017 Apr;21(2):175-181. doi: 10.1007/s10157-016-1286-4. Epub 2016 Jun 13. Clin Exp Nephrol. 2017. PMID: 27294581 Review.
-
Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase.Mol Cell Biochem. 2004 Sep;264(1-2):85-97. doi: 10.1023/b:mcbi.0000044378.09409.b5. Mol Cell Biochem. 2004. PMID: 15544038 Review.
-
The role of statin therapy in the management of cardiomyopathies.Curr Atheroscler Rep. 2009 Mar;11(2):118-23. doi: 10.1007/s11883-009-0019-5. Curr Atheroscler Rep. 2009. PMID: 19228485 Review.
-
Contribution of oxidative stress to pulmonary arterial hypertension.World J Cardiol. 2010 Oct 26;2(10):316-24. doi: 10.4330/wjc.v2.i10.316. World J Cardiol. 2010. PMID: 21160609 Free PMC article.
References
-
- Frohlich ED. Cardiac hypertrophy in hypertension. N Engl J Med. 1987;317:831–833. - PubMed
-
- Vasan RS, Levy D. The role of hypertension in the pathogenesis of heart failure. A clinical mechanistic overview. Arch Intern Med. 1996;156:1789–1796. - PubMed
-
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. - PubMed
-
- Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J. 1991;5:3037–3046. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
