

Motif-based fold assignment
- PMID: 11714913
- PMCID: PMC2374048
- DOI: 10.1110/ps.14401
Motif-based fold assignment
Abstract
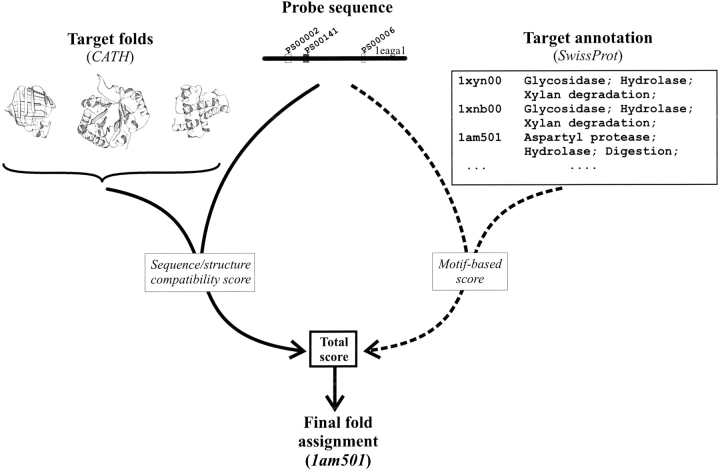
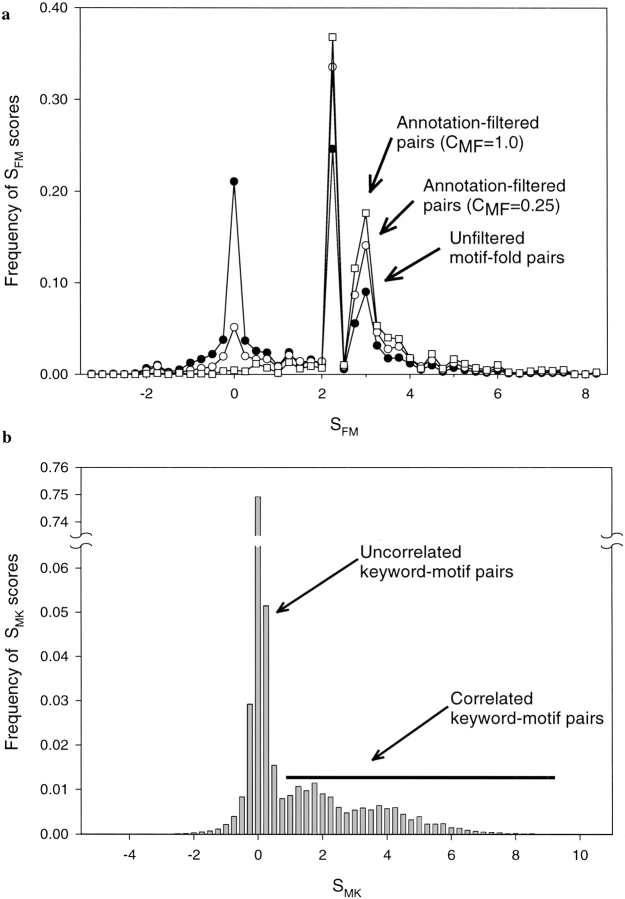
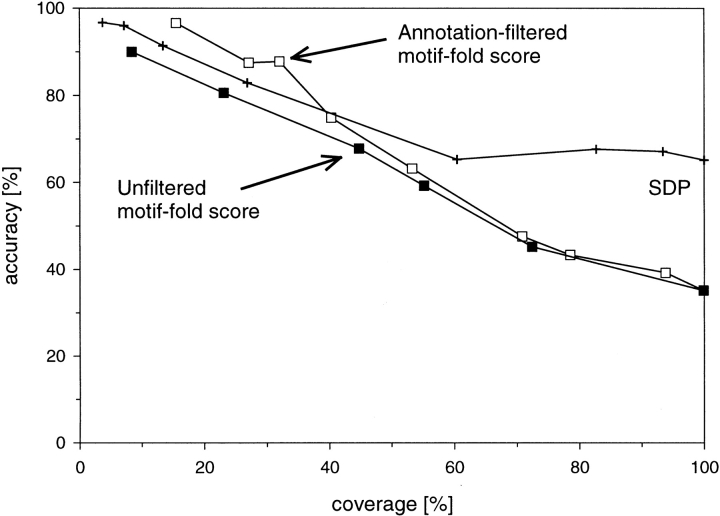
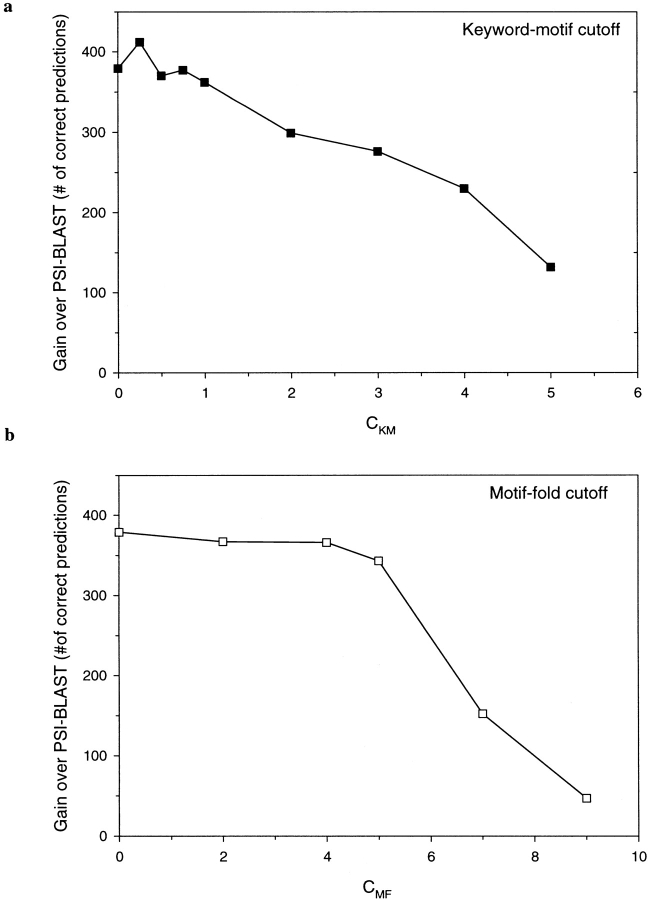
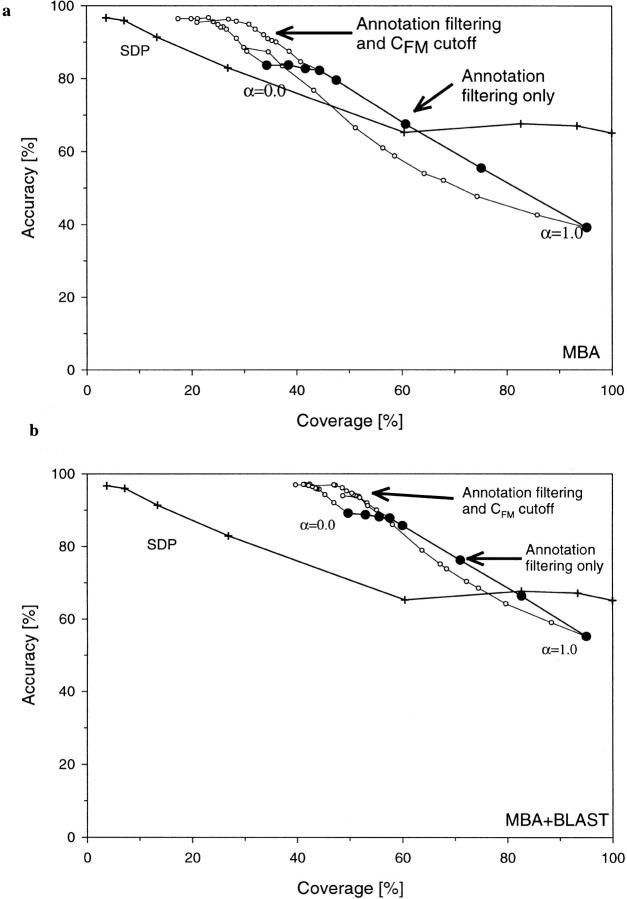
Conventional fold recognition techniques rely mainly on the analysis of the entire sequence of a protein. We present an MBA method to improve performance of any conventional sequence-based fold assignment. The method uses sequence motifs, such as those defined in the Prosite database, and the SwissProt annotation of the fold library. When combined with a simple SDP method, the coverage of MBA is comparable to the results obtained with PSI-BLAST. However, the set of the MBA predictions is significantly different from that of PSI-BLAST, leading to a 40% increase of the coverage for the combined MBA/PSI-BLAST method. The MBA approach can be easily adopted to include the results of sequence-independent function prediction methods and alternative motif and annotation databases. The method is available through the web server localized at http://www.doe-mbi.ucla.edu/mba.
Figures
References
-
- Andrade, M., Casari, G., de Daruvar, A., Sander, C., Schneider, R., Tamames, J., Valencia, A., and Ouzounis, C. 1997. Sequence analysis of the Methanococcus jannaschii genome and the prediction of protein function. Comput. Appl. Biosci. 13 481–483. - PubMed
-
- Andrade, M.A., Brown, N.P., Leroy, C., Hoersch, S., de Daruvar, A., Reich, C., Franchini, A., Tamames, J., Valencia, A., Ouzounis, C., and Sander, C. 1999. Automated genome sequence analysis and annotation. Bioinformatics 15 391–412. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
