

Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease
- PMID: 11717358
- PMCID: PMC6763929
- DOI: 10.1523/JNEUROSCI.21-23-09246.2001
Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease
Abstract
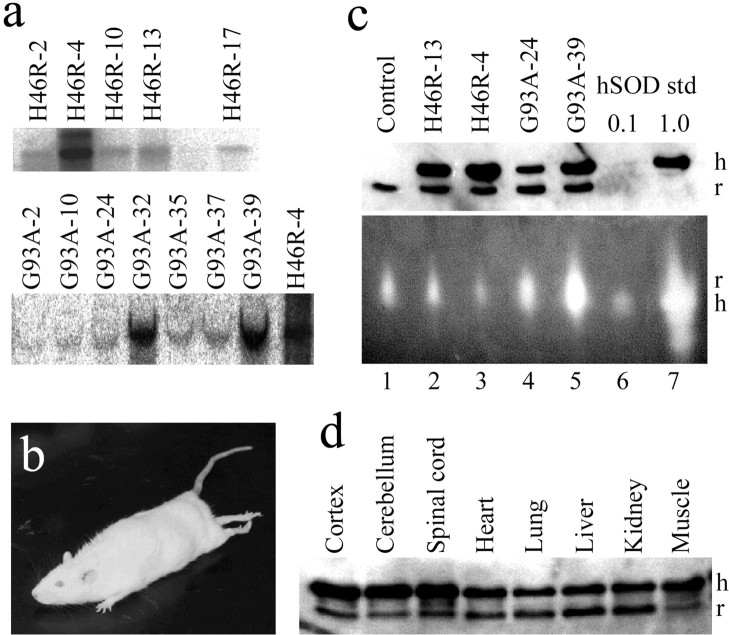
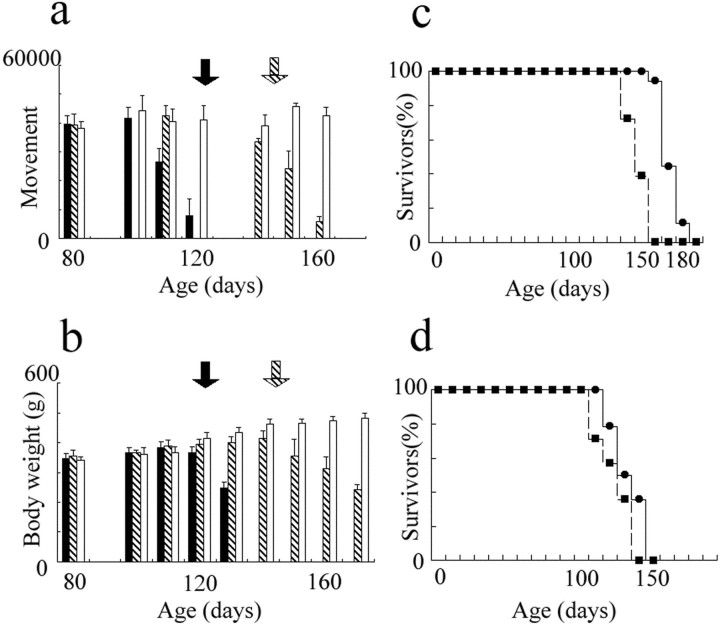
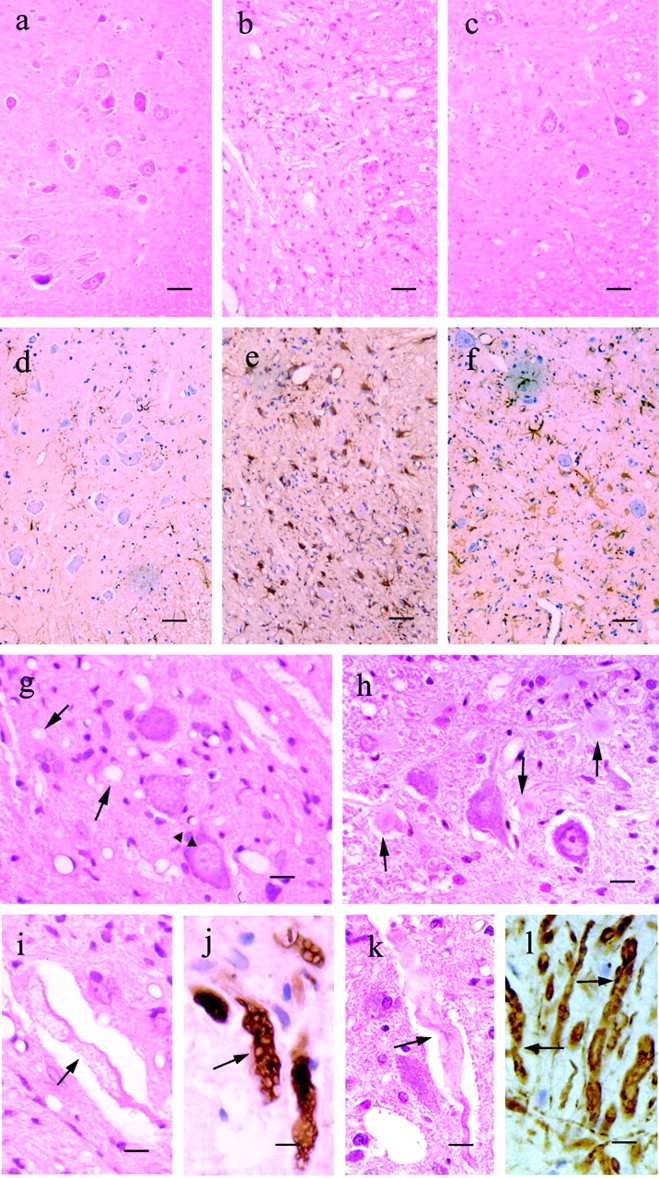
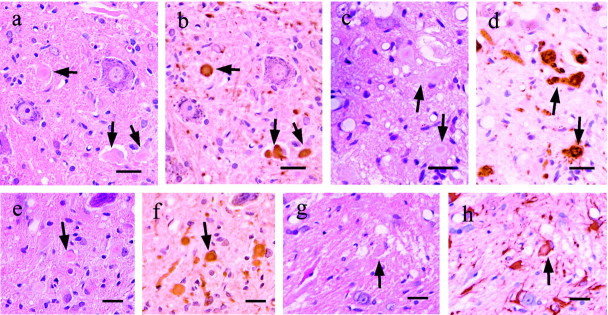
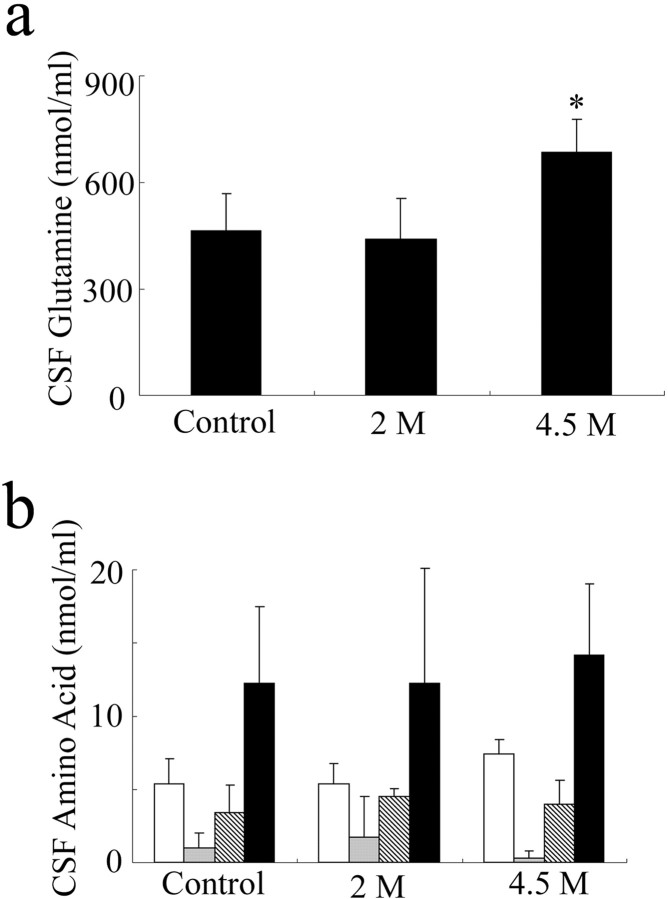
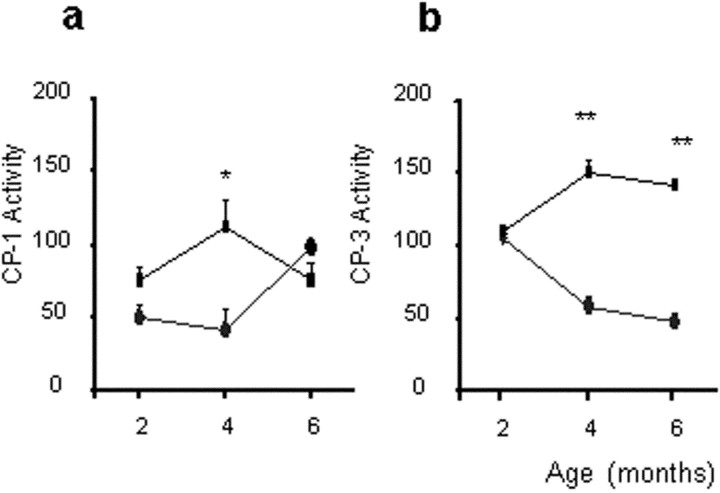
Some cases of familial amyotrophic lateral sclerosis (ALS) are caused by mutations in the gene encoding cytosolic, copper-zinc superoxide dismutase (SOD1). We report here that rats that express a human SOD1 transgene with two different ALS-associated mutations (G93A and H46R) develop striking motor neuron degeneration and paralysis. As in the human disease and transgenic ALS mice, pathological analysis demonstrates selective loss of motor neurons in the spinal cords of these transgenic rats. In spinal cord tissues, this is accompanied by activation of apoptotic genes known to be activated by mutant SOD1 protein in vitro and in vivo. These animals provide additional support for the proposition that motor neuron death in SOD1-related ALS reflects one or more acquired, neurotoxic properties of the mutant SOD1 protein. The larger size of this rat model as compared with the ALS mice will facilitate studies involving manipulations of spinal fluid (implantation of intrathecal catheters for chronic therapeutic studies; CSF sampling) and spinal cord (e.g., direct administration of viral- and cell-mediated therapies).
Figures
References
-
- Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Mild ALS in Japan associated with novel SOD mutation. Nat Genet. 1993;5:323–324. - PubMed
-
- Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Familial amyotrophic lateral sclerosis (ALS) in Japan associated with H46R mutation in Cu/Zn superoxide dismutase gene: a possible new subtype of familial ALS. J Neurol Sci. 1994;126:77–83. - PubMed
-
- Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. - PubMed
-
- Brown RHJ. Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell. 1995;80:687–692. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous