

Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms
- PMID: 11741196
- PMCID: PMC448439
- DOI: 10.1086/338446
Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms
Erratum in
- Am J Hum Genet. 2006 Jan;78(1):174
Abstract
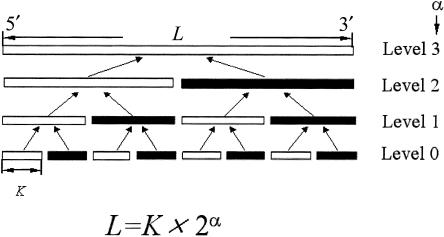
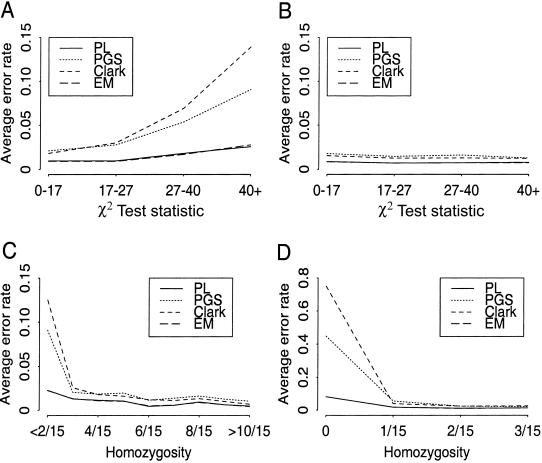
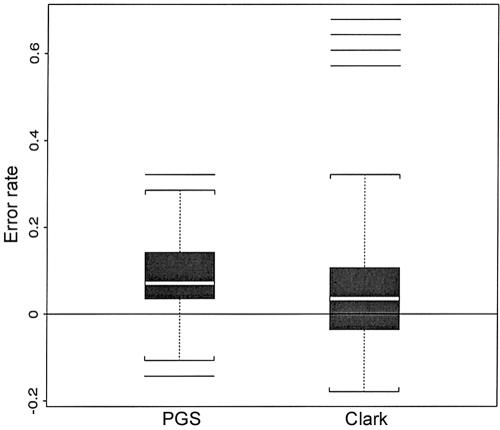
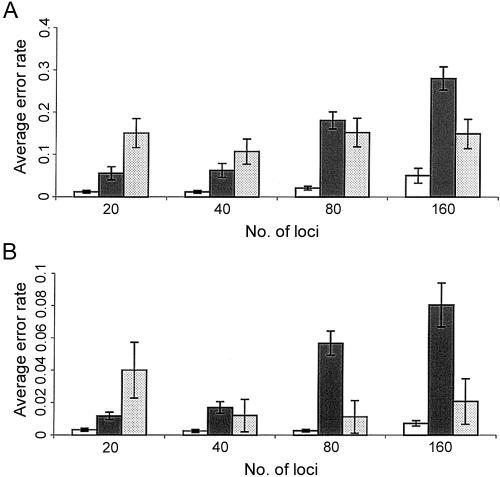
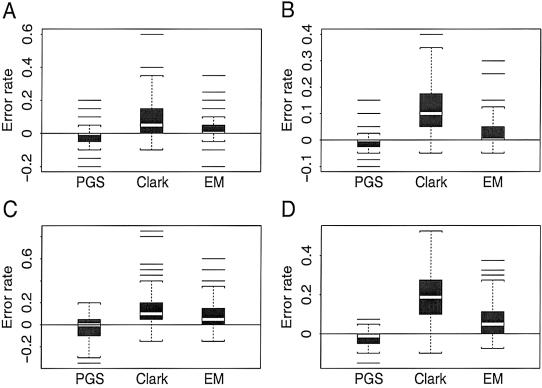
Haplotypes have gained increasing attention in the mapping of complex-disease genes, because of the abundance of single-nucleotide polymorphisms (SNPs) and the limited power of conventional single-locus analyses. It has been shown that haplotype-inference methods such as Clark's algorithm, the expectation-maximization algorithm, and a coalescence-based iterative-sampling algorithm are fairly effective and economical alternatives to molecular-haplotyping methods. To contend with some weaknesses of the existing algorithms, we propose a new Monte Carlo approach. In particular, we first partition the whole haplotype into smaller segments. Then, we use the Gibbs sampler both to construct the partial haplotypes of each segment and to assemble all the segments together. Our algorithm can accurately and rapidly infer haplotypes for a large number of linked SNPs. By using a wide variety of real and simulated data sets, we demonstrate the advantages of our Bayesian algorithm, and we show that it is robust to the violation of Hardy-Weinberg equilibrium, to the presence of missing data, and to occurrences of recombination hotspots.
Figures

Comment in
-
Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms.Am J Hum Genet. 2002 Nov;71(5):1242-7. doi: 10.1086/344207. Am J Hum Genet. 2002. PMID: 12452179 Free PMC article. No abstract available.
Similar articles
-
Haplotype reconstruction for diploid populations.Hum Hered. 2005;59(3):144-56. doi: 10.1159/000085938. Epub 2005 May 26. Hum Hered. 2005. PMID: 15925893
-
Inference of missing SNPs and information quantity measurements for haplotype blocks.Bioinformatics. 2005 May 1;21(9):2001-7. doi: 10.1093/bioinformatics/bti261. Epub 2005 Feb 4. Bioinformatics. 2005. PMID: 15699029
-
Haplotype block partitioning and tag SNP selection using genotype data and their applications to association studies.Genome Res. 2004 May;14(5):908-16. doi: 10.1101/gr.1837404. Epub 2004 Apr 12. Genome Res. 2004. PMID: 15078859 Free PMC article.
-
Algorithms for inferring haplotypes.Genet Epidemiol. 2004 Dec;27(4):334-47. doi: 10.1002/gepi.20024. Genet Epidemiol. 2004. PMID: 15368348 Review.
-
Haplotyping methods for pedigrees.Hum Hered. 2009;67(4):248-66. doi: 10.1159/000194978. Epub 2009 Jan 27. Hum Hered. 2009. PMID: 19172084 Free PMC article. Review.
Cited by
-
Inferring haplotypes of copy number variations from high-throughput data with uncertainty.G3 (Bethesda). 2011 Jun;1(1):35-42. doi: 10.1534/g3.111.000174. Epub 2011 Jun 1. G3 (Bethesda). 2011. PMID: 22384316 Free PMC article.
-
Haplotypic structure of the X chromosome in the COGA population sample and the quality of its reconstruction by extant software packages.BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S77. doi: 10.1186/1471-2156-6-S1-S77. BMC Genet. 2005. PMID: 16451691 Free PMC article.
-
Single nucleotide polymorphisms and haplotypes of the genes encoding the CYP1B1 in Korean women: no association with advanced endometriosis.J Assist Reprod Genet. 2007 Jul;24(7):271-7. doi: 10.1007/s10815-007-9122-0. Epub 2007 Jun 12. J Assist Reprod Genet. 2007. PMID: 17562158 Free PMC article.
-
A dynamic programming algorithm for haplotype block partitioning.Proc Natl Acad Sci U S A. 2002 May 28;99(11):7335-9. doi: 10.1073/pnas.102186799. Proc Natl Acad Sci U S A. 2002. PMID: 12032283 Free PMC article.
-
Polymorphisms within the canine MLPH gene are associated with dilute coat color in dogs.BMC Genet. 2005 Jun 16;6:34. doi: 10.1186/1471-2156-6-34. BMC Genet. 2005. PMID: 15960853 Free PMC article.
References
Electronic-Database Information
-
- Jun Liu's Home Page, http://www.people.fas.harvard.edu/~junliu/ (for example data files and documentation for HAPLOTYPER, EM-DeCODER, and HaplotypeManager)
-
- Long Lab, http://hjmuller.bio.uci.edu/~labhome/coalescent.html (for coalescent-process tools)
-
- Mathematics Genetics Group, http://www.stats.ox.ac.uk/mathgen/software.html (for PHASE)
References
-
- Akey J, Jin L, Xiong M (2001) Haplotypes vs single marker linkage disequilibrium tests: what do we gain? Eur J Hum Genet 9:291–300 - PubMed
-
- Chen R, Liu JS (1996) Predictive updating methods with application to Bayesian classification. J R Stat Soc Ser B 58:397–415
-
- Chiano MN, Clayton DG (1998) Fine genetic mapping using haplotype analysis and the missing data problem. Ann Hum Genet 62:55–60 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
