

Transforming growth factor-beta induction of smooth muscle cell phenotpye requires transcriptional and post-transcriptional control of serum response factor
- PMID: 11741973
- PMCID: PMC4421896
- DOI: 10.1074/jbc.M106649200
Transforming growth factor-beta induction of smooth muscle cell phenotpye requires transcriptional and post-transcriptional control of serum response factor
Abstract
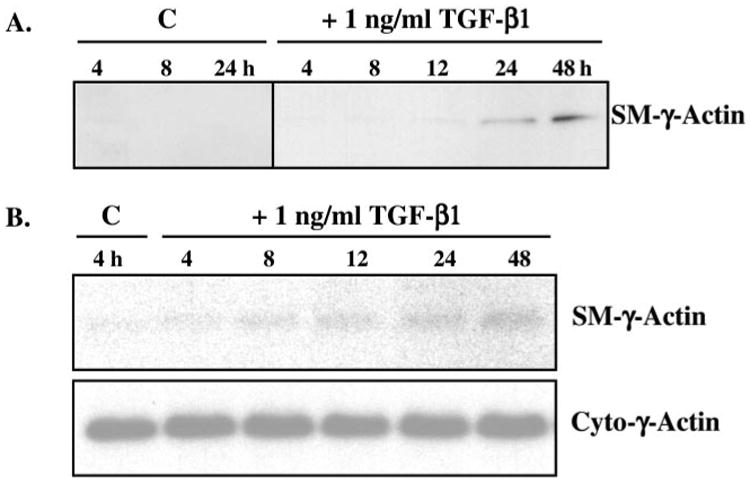
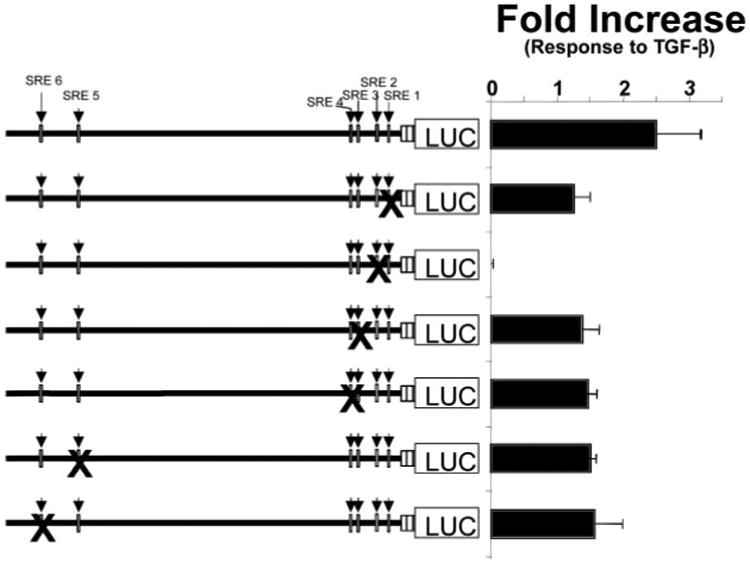
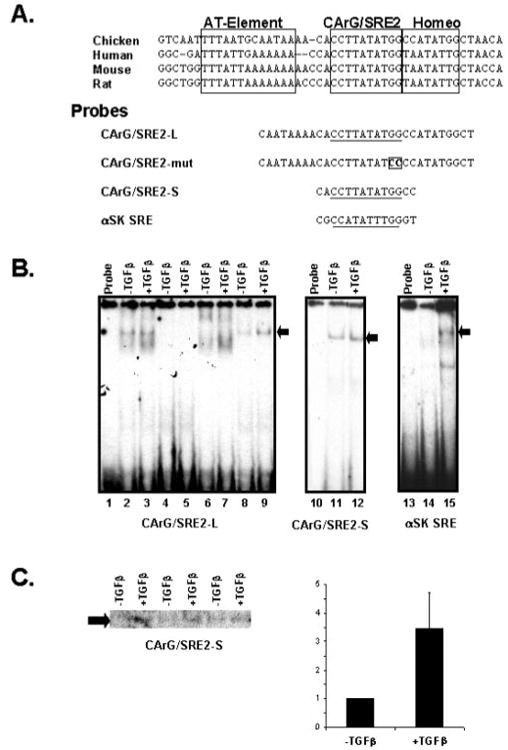
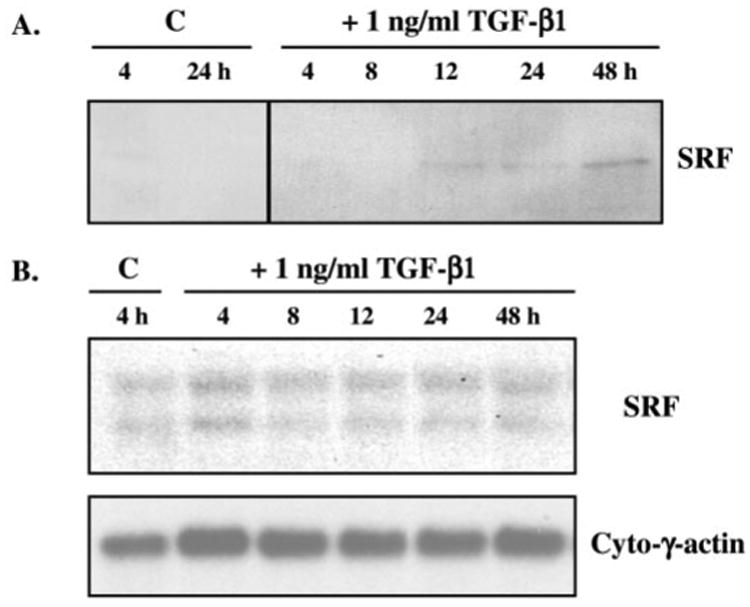
Transforming growth factor-beta induces a smooth muscle cell phenotype in undifferentiated mesenchymal cells. To elucidate the mechanism(s) of this phenotypic induction, we focused on the molecular regulation of smooth muscle-gamma-actin, whose expression is induced at late stages of smooth muscle differentiation and developmentally restricted to this lineage. Transforming growth factor-beta induced smooth muscle-gamma-actin protein, cytoskeletal localization, and mRNA expression in mesenchymal cells. Smooth muscle-gamma-actin promoter-luciferase reporter activity was enhanced by transforming growth factor-beta, and deletion analysis revealed that CArG box 2 in the promoter was necessary for this transcriptional activation. CArG motifs bind transcriptional activator serum response factor; gel shift analyses revealed increased binding of serum response factor-containing complexes to this site in response to transforming growth factor-beta, paralleled by increased serum response factor protein expression. Serum response factor expression was found to be up-regulated by transforming growth factor-beta via transcriptional activation of the gene and post-transcriptional regulation. Using mesenchymal cells stably transfected with wild type or dominant-negative serum response factor, we demonstrated that its expression is sufficient for induction of a smooth muscle phenotype in mesenchymal cells and is necessary for transforming growth factor-beta-mediated smooth muscle induction.
Figures



References
-
- Massaque J. Annu Rev Biochem. 1998;67:753–791. - PubMed
-
- Staehling-Hampton K, Hoffmann FM, Baylies MK, Rushton E, Bate M. Nature. 1994;372:783–786. - PubMed
-
- Roberts A. Wound Repair Regen. 1995;3:408–418. - PubMed
-
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Development (Camb) 1995;121:1845–1854. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
