

Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo
- PMID: 11756516
- PMCID: PMC6757602
- DOI: 10.1523/JNEUROSCI.22-01-00324.2002
Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo
Abstract
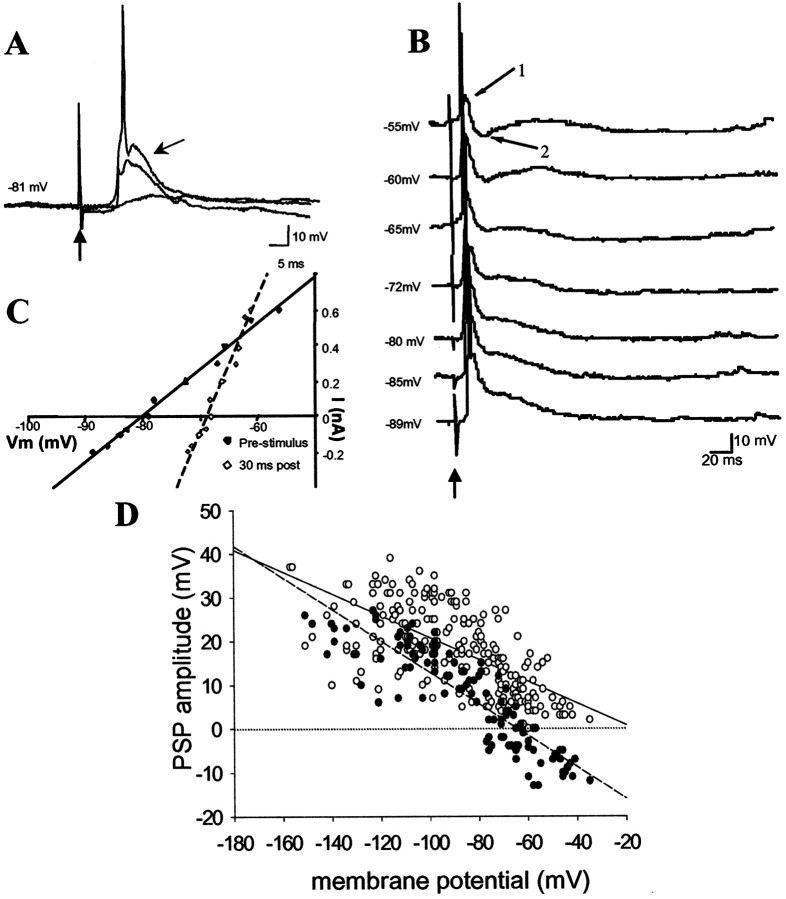
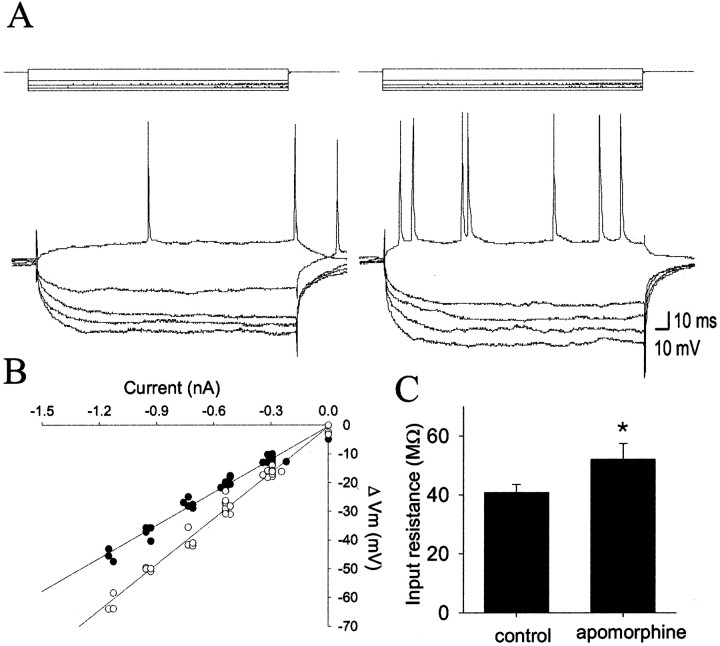
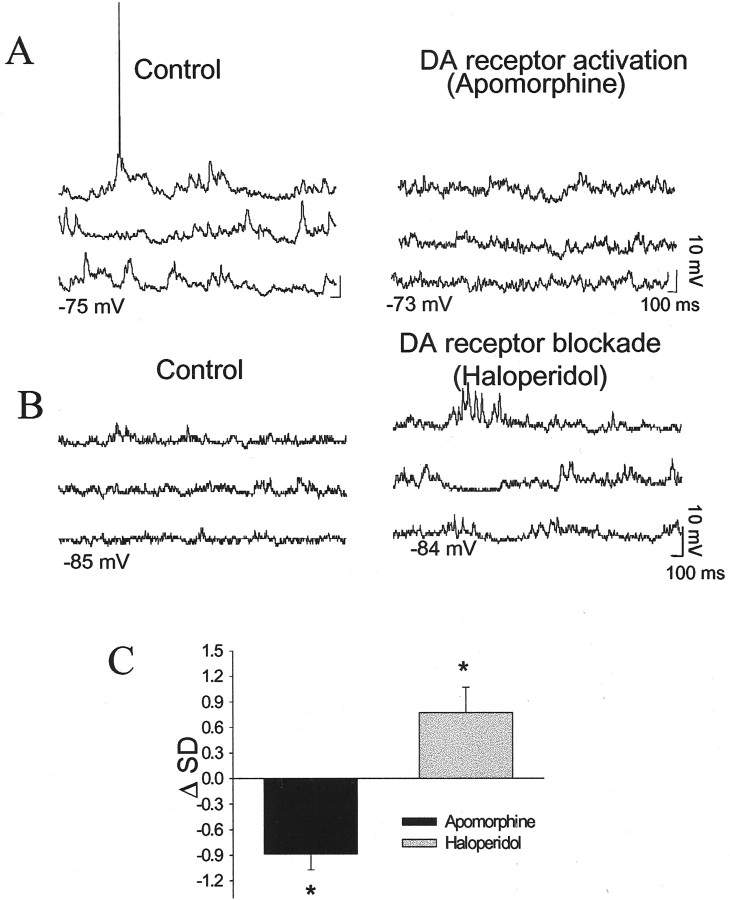
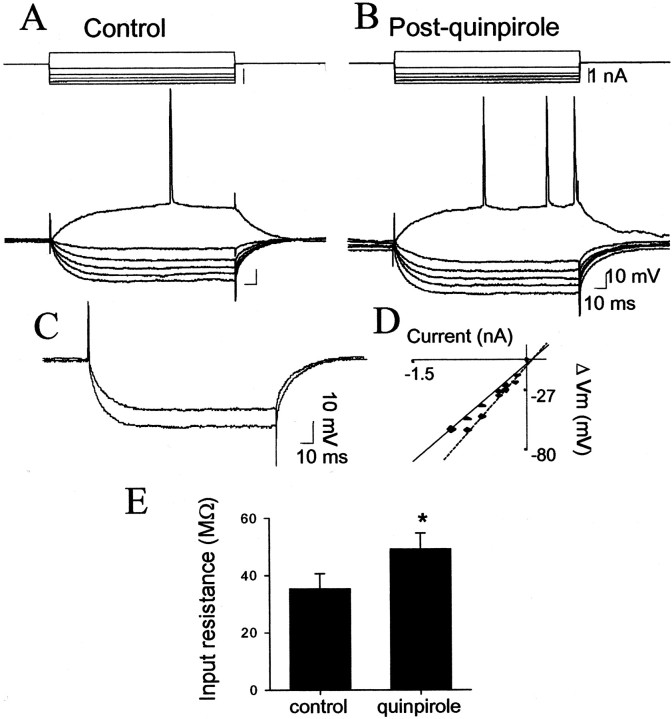
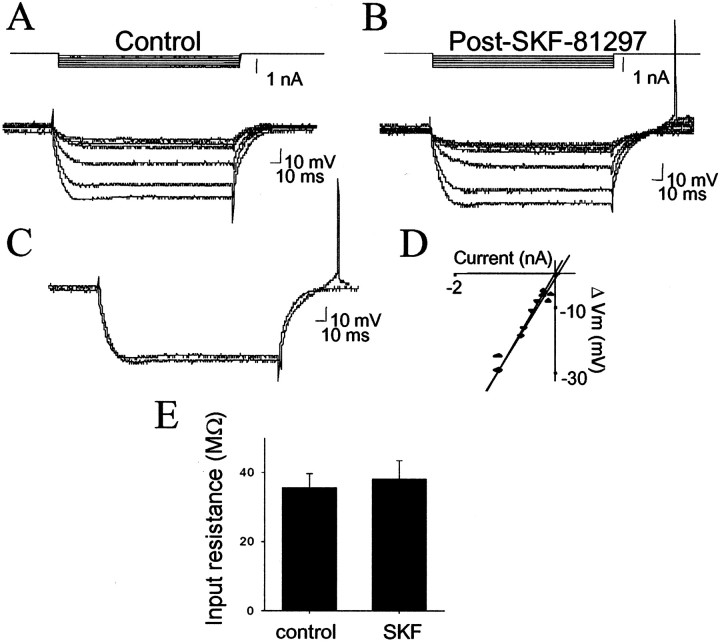
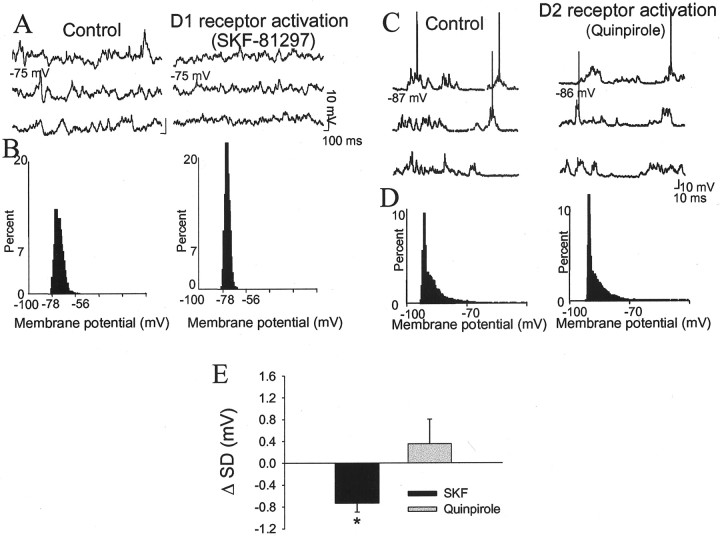
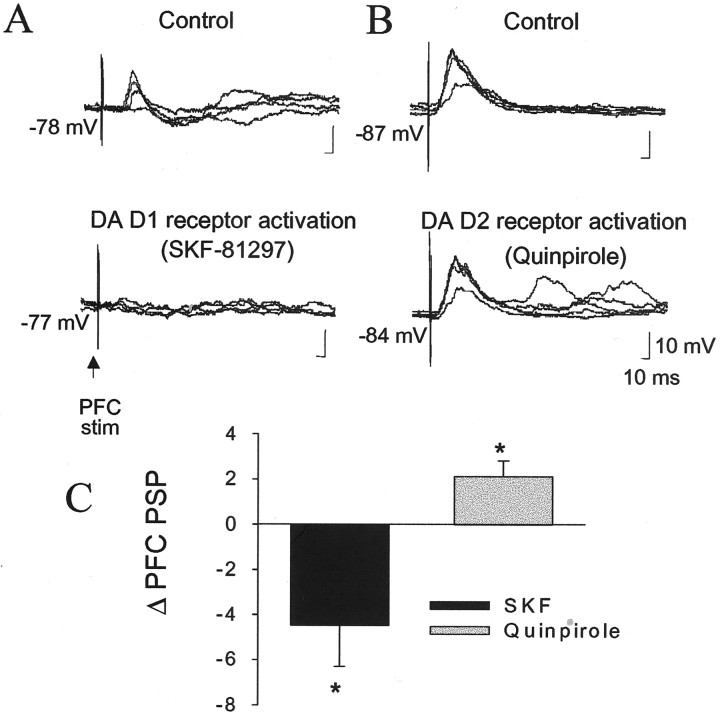
The basolateral amygdala (BLA) is believed to be involved in schizophrenia, depression, and other disorders that display affective components. The neuronal activity of the BLA, and BLA-mediated affective behaviors, are driven by sensory stimuli transmitted in part from sensory association cortical regions. These same behaviors may be regulated by prefrontal cortical (PFC) inputs to the BLA. However, it is unclear how two sets of glutamatergic inputs to the BLA can impose opposing actions on BLA-mediated behaviors; specifically, it is unclear how PFC inputs exert inhibitory actions over BLA projection neurons. Dopamine (DA) receptor activation enhances BLA-mediated behaviors. Although we have demonstrated that DA suppresses medial PFC inputs to the BLA and enhances sensory cortical inputs, the precise cellular mechanisms for its actions are unknown. In this study we use in vivo intracellular recordings to determine the means by which glutamatergic inputs from the PFC inhibit BLA projection neurons, contrast that with glutamatergic inputs from the association sensory cortex (Te3) that drive BLA projection neurons, and examine the effects of DA receptor activation on neuronal excitability, spontaneous postsynaptic potentials (PSPs), and PFC-evoked PSPs. We found that PFC stimulation inhibits BLA projection neurons by three mechanisms: chloride-mediated hyperpolarization, a persistent decrease in neuronal input resistance, and shunting of PSPs; all effects are possibly attributable to recruitment of inhibitory interneurons. DA receptor activation enhanced neuronal input resistance by a postsynaptic mechanism (via DA D2 receptors), suppressed spontaneously occurring and PFC-evoked PSPs (via DA D1 receptors), and enhanced Te3-evoked PSPs.
Figures
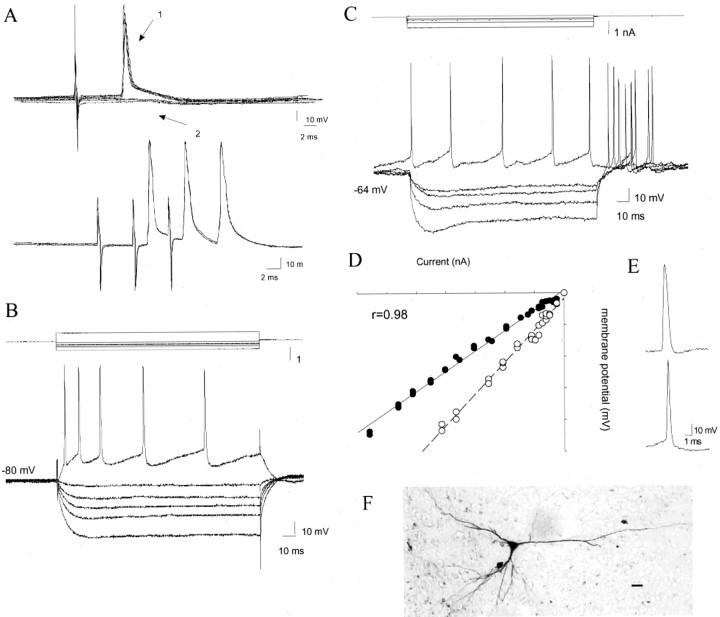
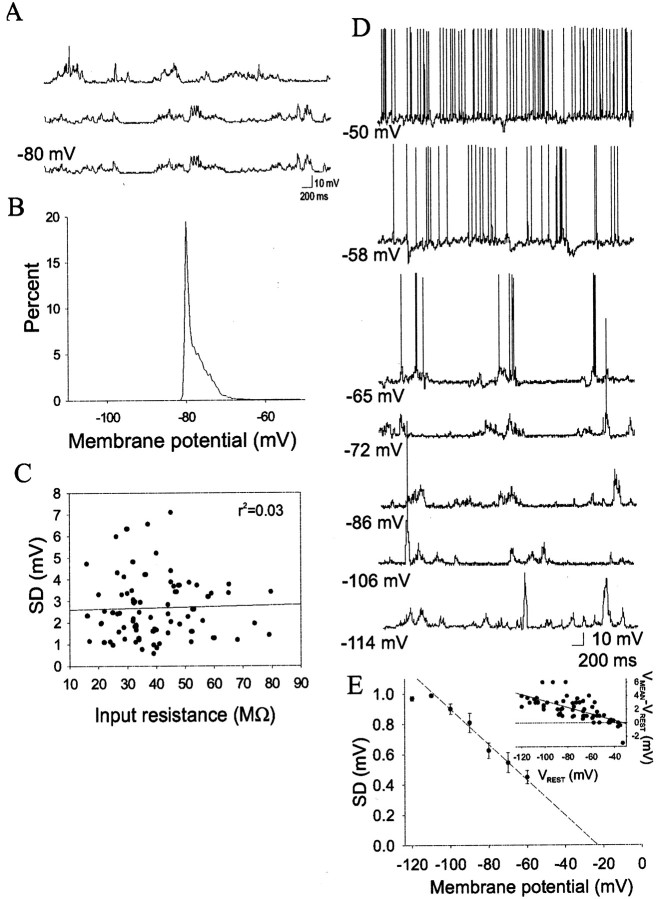
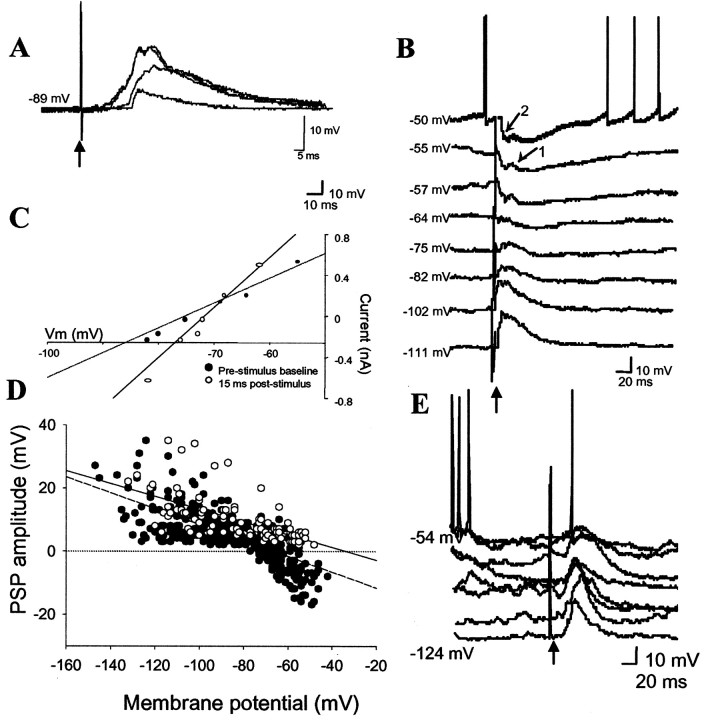
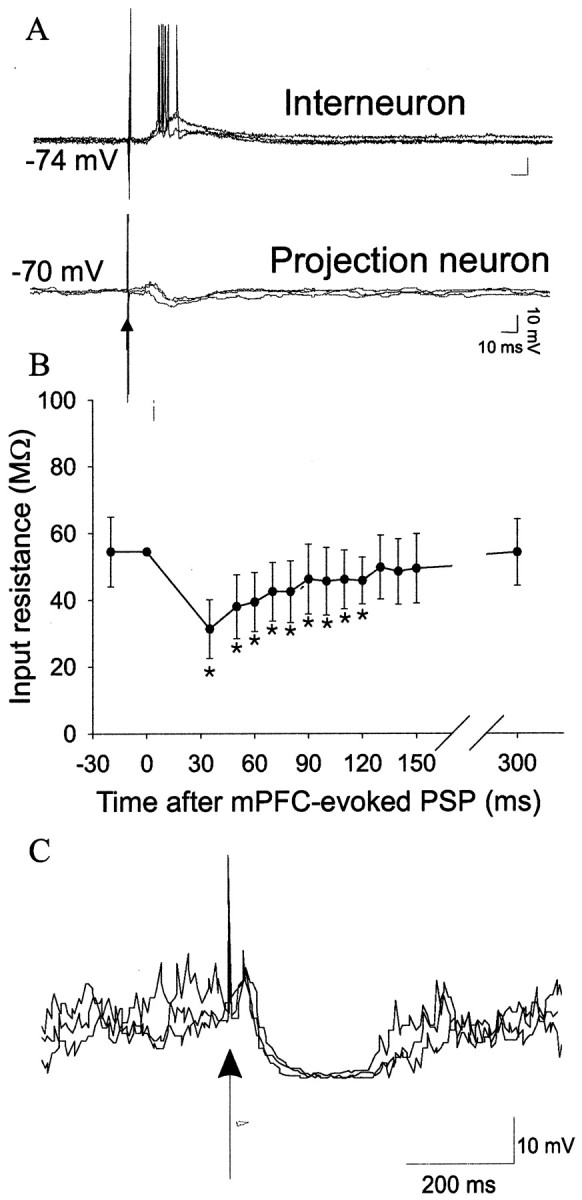
Similar articles
-
Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats.J Neurosci. 2001 Jun 1;21(11):4090-103. doi: 10.1523/JNEUROSCI.21-11-04090.2001. J Neurosci. 2001. PMID: 11356897 Free PMC article.
-
Dopamine modulates excitability of basolateral amygdala neurons in vitro.J Neurophysiol. 2005 Mar;93(3):1598-610. doi: 10.1152/jn.00843.2004. Epub 2004 Nov 10. J Neurophysiol. 2005. PMID: 15537813
-
Dopaminergic regulation of inhibitory and excitatory transmission in the basolateral amygdala-prefrontal cortical pathway.J Neurosci. 2007 Feb 21;27(8):2045-57. doi: 10.1523/JNEUROSCI.5474-06.2007. J Neurosci. 2007. PMID: 17314300 Free PMC article.
-
Regulation of conditioned responses of basolateral amygdala neurons.Physiol Behav. 2002 Dec;77(4-5):489-93. doi: 10.1016/s0031-9384(02)00909-5. Physiol Behav. 2002. PMID: 12526988 Review.
-
Dopamine's Actions in Primate Prefrontal Cortex: Challenges for Treating Cognitive Disorders.Pharmacol Rev. 2015 Jul;67(3):681-96. doi: 10.1124/pr.115.010512. Pharmacol Rev. 2015. PMID: 26106146 Free PMC article. Review.
Cited by
-
Alcohol Effects on Stress Pathways: Impact on Craving and Relapse Risk.Can J Psychiatry. 2016 Mar;61(3):145-53. doi: 10.1177/0706743716632512. Epub 2016 Feb 9. Can J Psychiatry. 2016. PMID: 27254089 Free PMC article. Review.
-
In vivo voltage-dependent influences on summation of synaptic potentials in neurons of the lateral nucleus of the amygdala.Neuroscience. 2012 Dec 13;226:101-18. doi: 10.1016/j.neuroscience.2012.09.012. Epub 2012 Sep 16. Neuroscience. 2012. PMID: 22989917 Free PMC article.
-
Organic cation transporter 3 (OCT3) is localized to intracellular and surface membranes in select glial and neuronal cells within the basolateral amygdaloid complex of both rats and mice.Brain Struct Funct. 2017 May;222(4):1913-1928. doi: 10.1007/s00429-016-1315-9. Epub 2016 Sep 22. Brain Struct Funct. 2017. PMID: 27659446 Free PMC article.
-
Memory enhancement induced by post-training intrabasolateral amygdala infusions of beta-adrenergic or muscarinic agonists requires activation of dopamine receptors: Involvement of right, but not left, basolateral amygdala.Learn Mem. 2005 Sep-Oct;12(5):527-32. doi: 10.1101/lm.97405. Learn Mem. 2005. PMID: 16204205 Free PMC article.
-
Altered prelimbic cortex output during cue-elicited drug seeking.J Neurosci. 2004 Aug 4;24(31):6889-97. doi: 10.1523/JNEUROSCI.1685-04.2004. J Neurosci. 2004. PMID: 15295023 Free PMC article.
References
-
- Alheid G, de Olmos JS, Beltramino CA. Amygdala and extended amygdala. In: Paxinos G, editor. The rat nervous system, Ed 2. Academic; Sydney: 1995. pp. 495–572.
-
- Al Maskati HA, Zbrozyna AW. Stimulation in prefrontal cortex area inhibits cardiovascular and motor components of the defense reaction in rats. J Auton Nerv Syst. 1989;28:117–126. - PubMed
-
- Anderson JS, Carandini M, Ferster D. Orientation tuning of input conductance, excitation, and inhibition in cat primary visual cortex. J Neurophysiol. 2000;84:909–926. - PubMed
-
- Angrist BM, Gershon S. The phenomenology of experimentally induced amphetamine psychosis: preliminary observations. Biol Psychiatry. 1970;2:95–107. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous