

Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements
- PMID: 11756544
- PMCID: PMC139739
- DOI: 10.1128/MCB.22.2.480-491.2002
Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements
Abstract
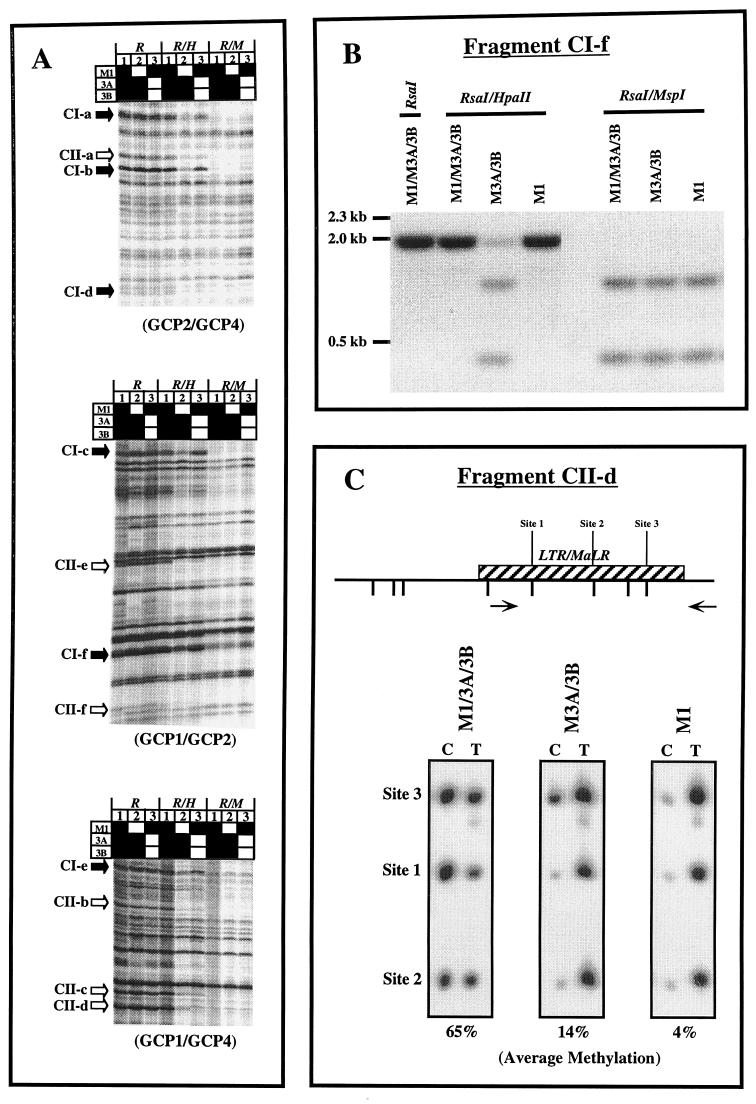
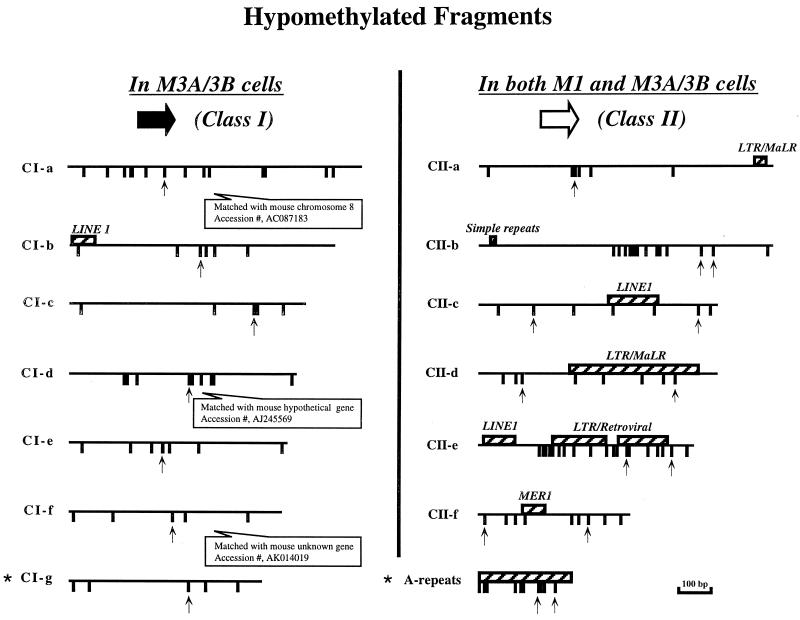
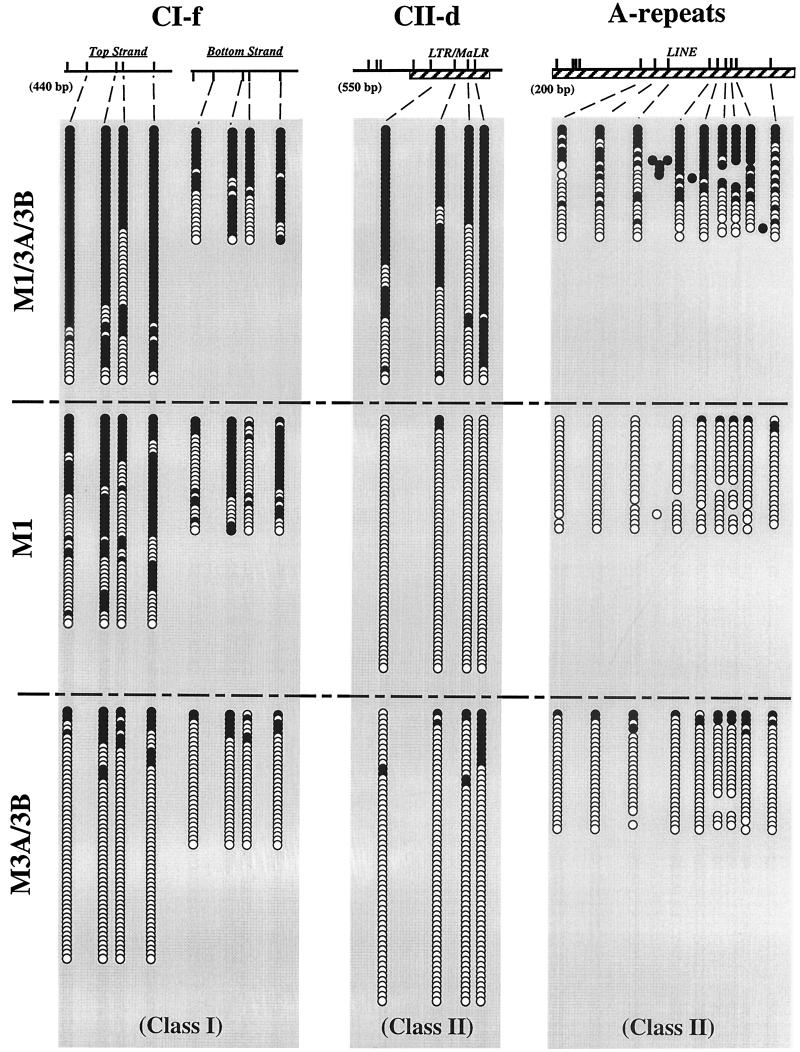
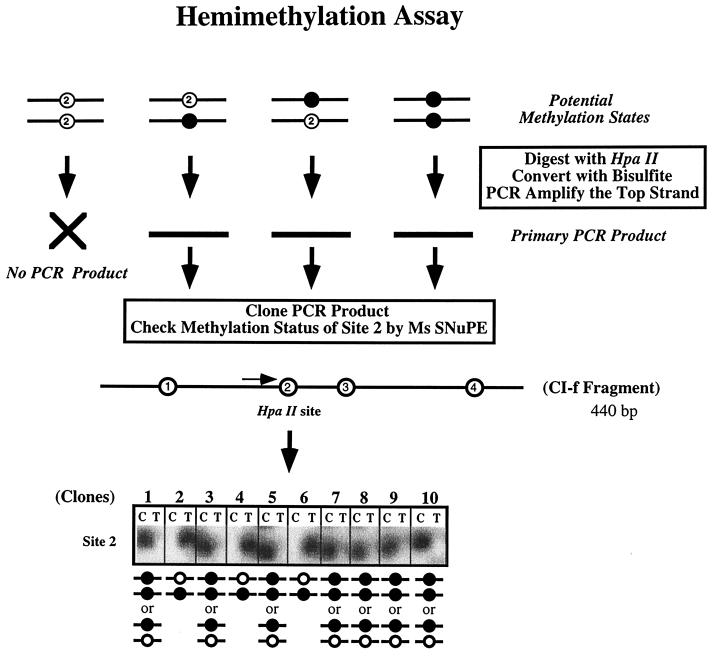
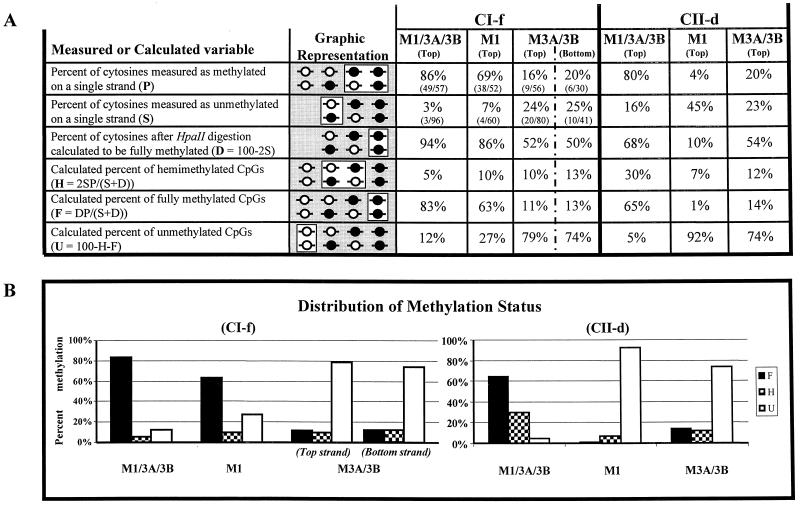
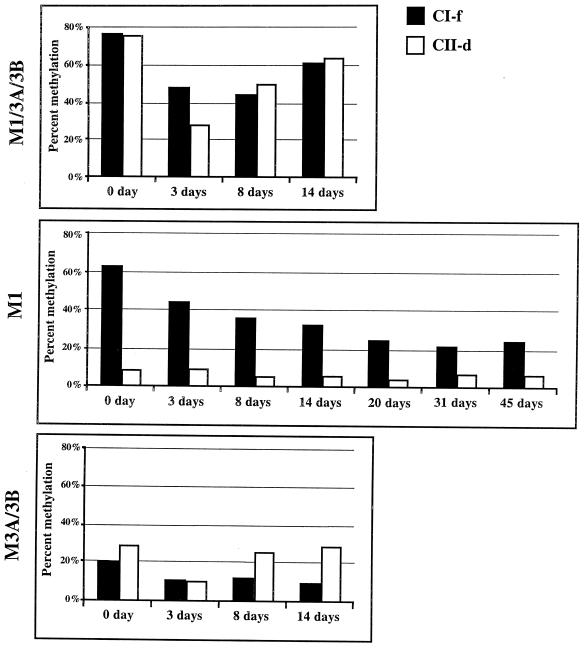
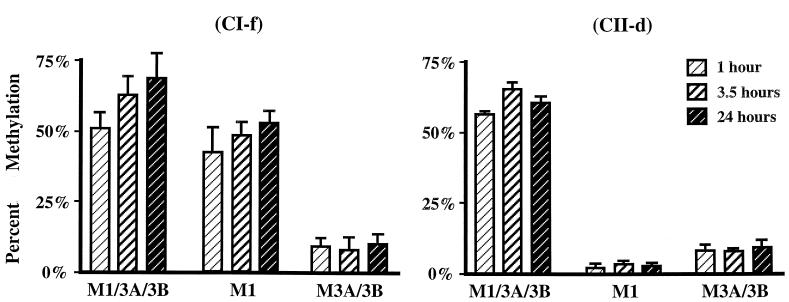
We used mouse embryonic stem (ES) cells with systematic gene knockouts for DNA methyltransferases to delineate the roles of DNA methyltransferase 1 (Dnmt1) and Dnmt3a and -3b in maintaining methylation patterns in the mouse genome. Dnmt1 alone was able to maintain methylation of most CpG-poor regions analyzed. In contrast, both Dnmt1 and Dnmt3a and/or Dnmt3b were required for methylation of a select class of sequences which included abundant murine LINE-1 promoters. We used a novel hemimethylation assay to show that even in wild-type cells these sequences contain high levels of hemimethylated DNA, suggestive of poor maintenance methylation. We showed that Dnmt3a and/or -3b could restore methylation of these sequences to pretreatment levels following transient exposure of cells to 5-aza-CdR, whereas Dnmt1 by itself could not. We conclude that ongoing de novo methylation by Dnmt3a and/or Dnmt3b compensates for inefficient maintenance methylation by Dnmt1 of these endogenous repetitive sequences. Our results reveal a previously unrecognized degree of cooperativity among mammalian DNA methyltransferases in ES cells.
Figures
References
-
- Adams, R. L. 1971. The relationship between synthesis and methylation of DNA in mouse fibroblasts. Biochim. Biophys. Acta 254:205–212. - PubMed
-
- Baylin, S. B., and J. G. Herman. 2000. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 16:168–174. - PubMed
-
- Bird, A., M. Taggart, M. Frommer, O. J. Miller, and D. Macleod. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40:91–99. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases