

Mutations of the woodchuck hepatitis virus polymerase gene that confer resistance to lamivudine and 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil
- PMID: 11773397
- PMCID: PMC135858
- DOI: 10.1128/jvi.76.3.1213-1223.2002
Mutations of the woodchuck hepatitis virus polymerase gene that confer resistance to lamivudine and 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil
Abstract
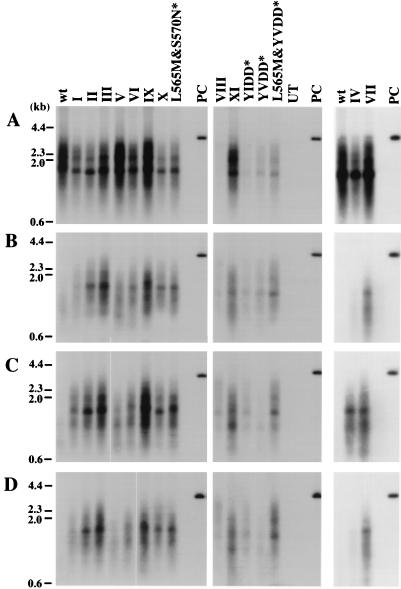
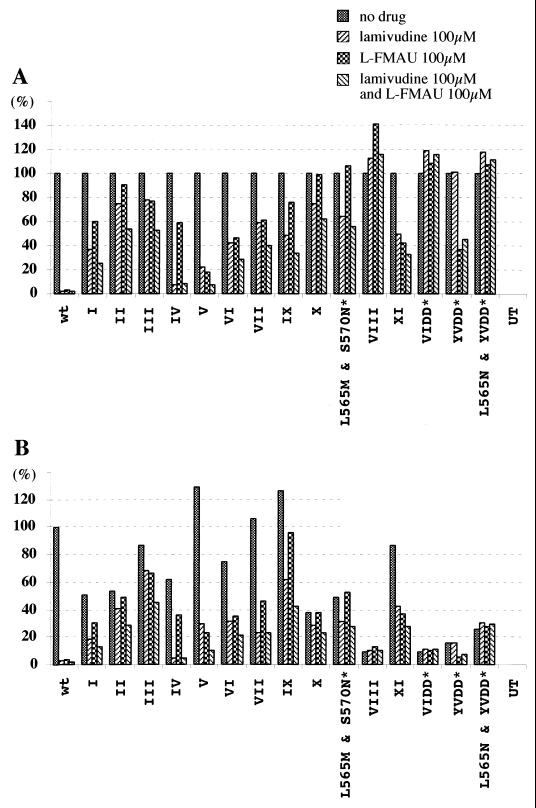
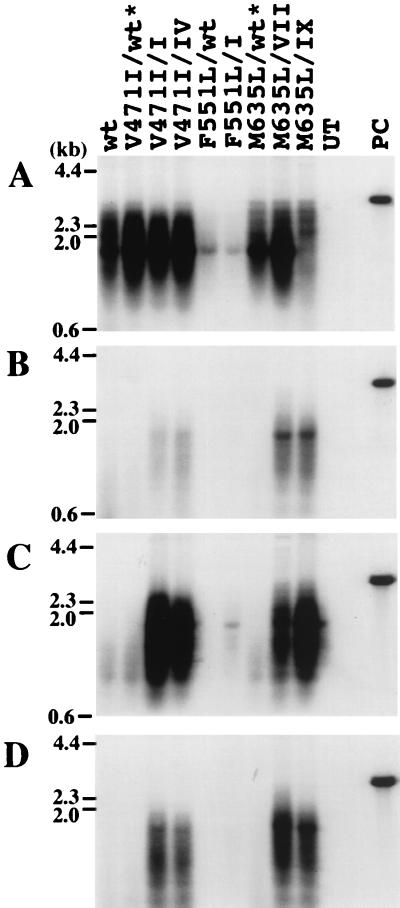
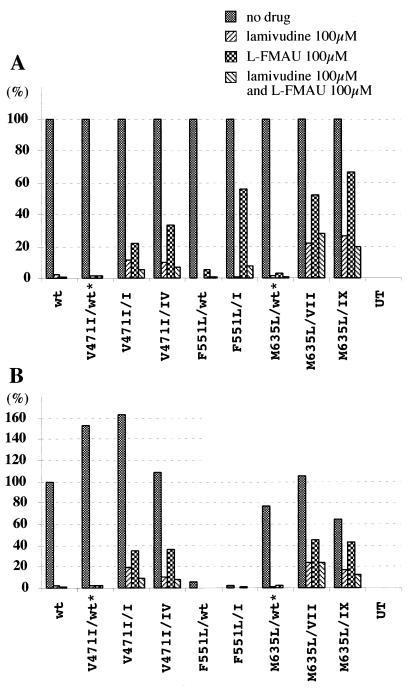
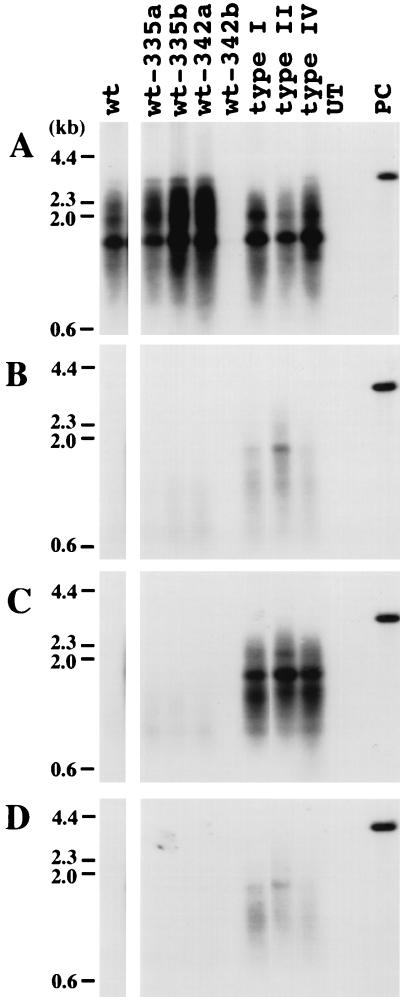
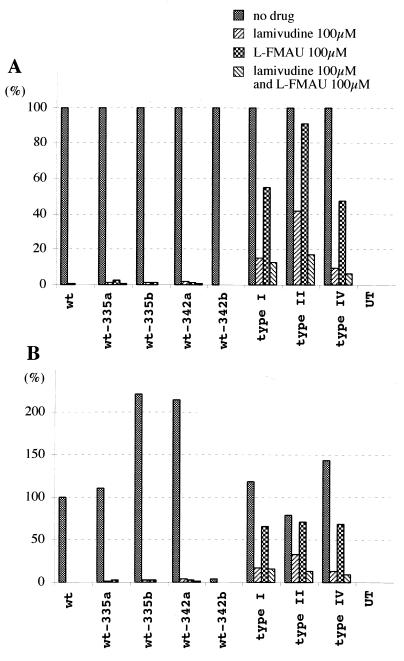
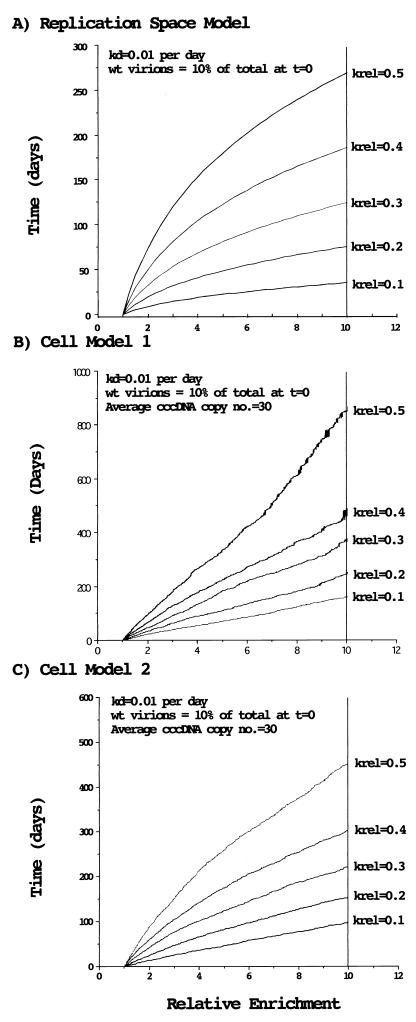
Administration of either lamivudine (2'-deoxy-3'-thiacytidine) or L-FMAU (2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil) to woodchucks chronically infected with woodchuck hepatitis virus (WHV) induces a transient decline in virus titers. However, within 6 to 12 months, virus titers begin to increase towards pretreatment levels. This is associated with the emergence of virus strains with mutations of the B and C regions of the viral DNA polymerase (T. Zhou et al., Antimicrob. Agents Chemother. 43:1947-1954, 1999; Y. Zhu et al., J. Virol. 75:311-322, 2001). The present study was carried out to determine which of the mutants that we have identified conferred resistance to lamivudine and/or to L-FMAU. When inserted into a laboratory strain of WHV, each of the mutations, or combinations of mutations, of regions B and C produced a DNA replication-competent virus and typically conferred resistance to both nucleoside analogs in cell culture. Sequencing of the polymerase active site also occasionally revealed other mutations, but these did not appear to contribute to drug resistance. Moreover, in transfected cells, most of the mutants synthesized viral DNA nearly as efficiently as wild-type WHV. Computational models suggested that persistence of several of the WHV mutants as prevalent species in the serum and, by inference, liver for up to 6 months following drug withdrawal required a replication efficiency of at least 10 to 30% of that of the wild type. However, their delayed emergence during therapy suggested replication efficiency in the presence of the drug that was still well below that of wild-type WHV in the absence of the drug.
Figures



Similar articles
-
Mutations in the conserved woodchuck hepatitis virus polymerase FLLA and YMDD regions conferring resistance to lamivudine.Antiviral Res. 2002 Jul;55(1):141-50. doi: 10.1016/s0166-3542(02)00019-0. Antiviral Res. 2002. PMID: 12076758
-
Emergence of drug-resistant populations of woodchuck hepatitis virus in woodchucks treated with the antiviral nucleoside lamivudine.Antimicrob Agents Chemother. 1999 Aug;43(8):1947-54. doi: 10.1128/AAC.43.8.1947. Antimicrob Agents Chemother. 1999. PMID: 10428918 Free PMC article.
-
Antiviral activity of clevudine [L-FMAU, (1-(2-fluoro-5-methyl-beta, L-arabinofuranosyl) uracil)] against woodchuck hepatitis virus replication and gene expression in chronically infected woodchucks (Marmota monax).Hepatology. 2001 Jan;33(1):254-66. doi: 10.1053/jhep.2001.20899. Hepatology. 2001. PMID: 11124844
-
Preclinical investigation of L-FMAU as an anti-hepatitis B virus agent.Antivir Ther. 1998;3(Suppl 3):113-21. Antivir Ther. 1998. PMID: 10726061 Review.
-
Resistance of hepatitis B virus to antiviral drugs: current aspects and directions for future investigation.Antivir Chem Chemother. 2001 Jan;12(1):1-35. doi: 10.1177/095632020101200101. Antivir Chem Chemother. 2001. PMID: 11437320 Review.
Cited by
-
In vitro drug susceptibility analysis of hepatitis B virus clinical quasispecies populations.J Clin Microbiol. 2007 Oct;45(10):3335-41. doi: 10.1128/JCM.00272-07. Epub 2007 Aug 8. J Clin Microbiol. 2007. PMID: 17687019 Free PMC article.
-
The Woodchuck, a Nonprimate Model for Immunopathogenesis and Therapeutic Immunomodulation in Chronic Hepatitis B Virus Infection.Cold Spring Harb Perspect Med. 2015 Oct 28;5(12):a021451. doi: 10.1101/cshperspect.a021451. Cold Spring Harb Perspect Med. 2015. PMID: 26511761 Free PMC article. Review.
-
Generation of stable cell lines expressing Lamivudine-resistant hepatitis B virus for antiviral-compound screening.Antimicrob Agents Chemother. 2003 Jun;47(6):1936-42. doi: 10.1128/AAC.47.6.1936-1942.2003. Antimicrob Agents Chemother. 2003. PMID: 12760870 Free PMC article.
-
Identification and characterization of clevudine-resistant mutants of hepatitis B virus isolated from chronic hepatitis B patients.J Virol. 2010 May;84(9):4494-503. doi: 10.1128/JVI.02066-09. Epub 2010 Feb 17. J Virol. 2010. PMID: 20164224 Free PMC article.
-
Discovery and development of anti-HBV agents and their resistance.Molecules. 2010 Aug 27;15(9):5878-908. doi: 10.3390/molecules15095878. Molecules. 2010. PMID: 20802402 Free PMC article. Review.
References
-
- Ahmed, S. N. S., D. Tavan, C. Pichoud, F. Berby, L. Stuyver, M. Johnson, P. Merle, H. Abidi, C. Trepo, and F. Zoulim. 2000. Early detection of viral resistance by determination of hepatitis B virus polymerase mutations in patients treated with lamivudine for chronic hepatitis B. Hepatology 32:1078–1088. - PubMed
-
- Allen, M. I., M. Deslauriers, C. W. Andrews, G. A. Tipples, K.-A. Walters, D. L. J. Tyrrell, N. Brown, and L. D. Condreay. 1998. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Hepatology 27:1670–1677. - PubMed
-
- Aye, T. T., A. Bartholomeusz, T. Shaw, S. Bowden, A. Breschkin, J. McMillan, P. Angus, and S. Locarnini. 1997. Hepatitis B virus polymerase mutations during antiviral therapy in a patient following liver transplantation. J. Hepatol. 26:1148–1153. - PubMed
-
- Bartholomeusz, A., L. C. Groenen, and S. A. Locarnini. 1997. Clinical experience with famciclovir against hepatitis B virus and development of resistance. Intervirology 40:337–342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
