

Relating whole-genome expression data with protein-protein interactions
- PMID: 11779829
- PMCID: PMC155252
- DOI: 10.1101/gr.205602
Relating whole-genome expression data with protein-protein interactions
Abstract
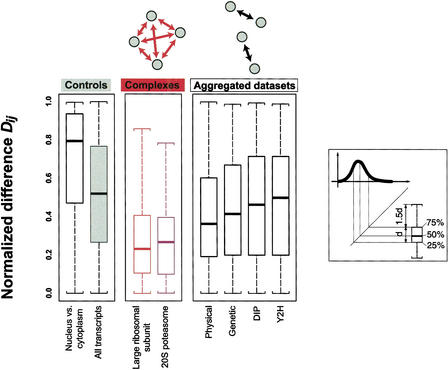
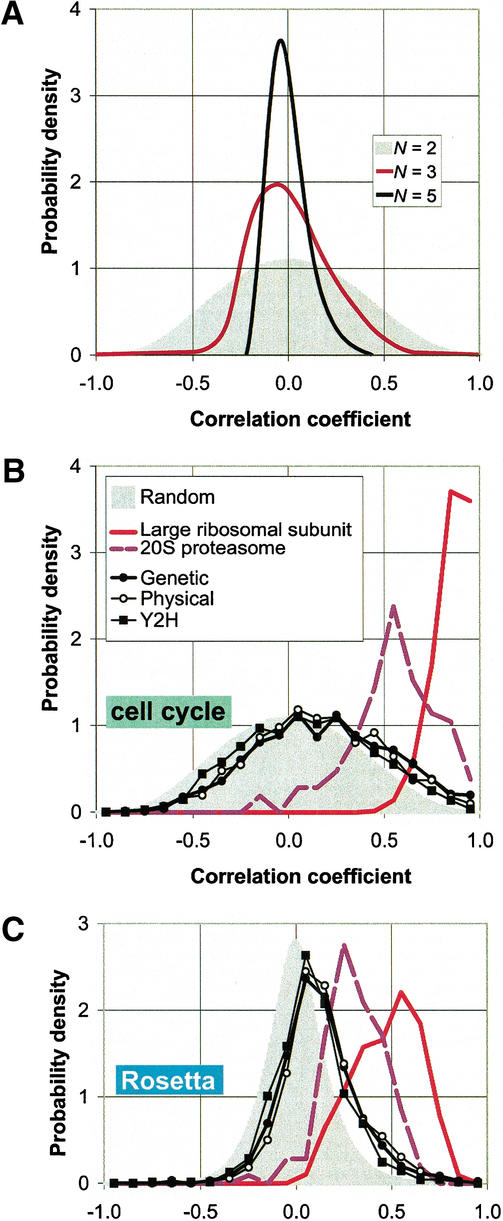
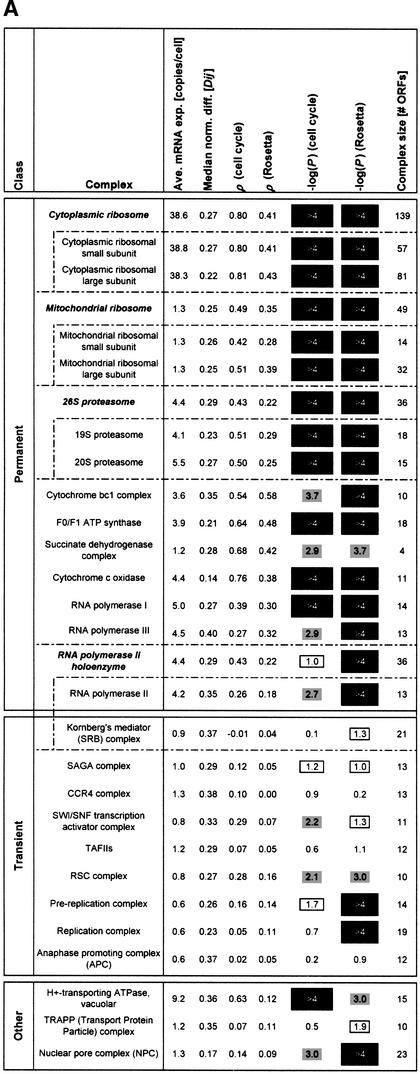
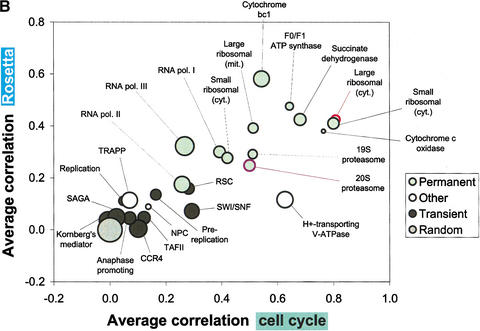
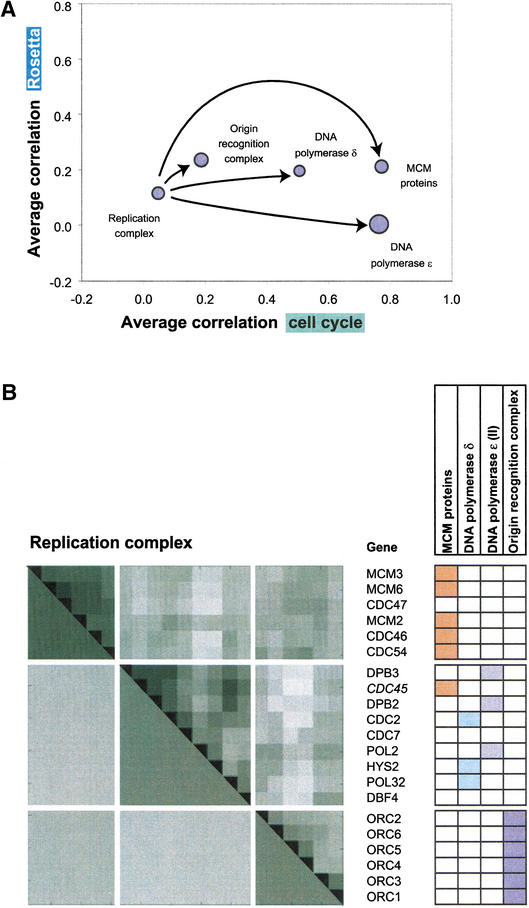
We investigate the relationship of protein-protein interactions with mRNA expression levels, by integrating a variety of data sources for yeast. We focus on known protein complexes that have clearly defined interactions between their subunits. We find that subunits of the same protein complex show significant coexpression, both in terms of similarities of absolute mRNA levels and expression profiles, e.g., we can often see subunits of a complex having correlated patterns of expression over a time course. We classify the yeast protein complexes as either permanent or transient, with permanent ones being maintained through most cellular conditions. We find that, generally, permanent complexes, such as the ribosome and proteasome, have a particularly strong relationship with expression, while transient ones do not. However, we note that several transient complexes, such as the RNA polymerase II holoenzyme and the replication complex, can be subdivided into smaller permanent ones, which do have a strong relationship to gene expression. We also investigated the interactions in aggregated, genome-wide data sets, such as the comprehensive yeast two-hybrid experiments, and found them to have only a weak relationship with gene expression, similar to that of transient complexes. (Further details on genecensus.org/expression/interactions and bioinfo.mbb.yale.edu/expression/interactions.)
Figures
References
-
- Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 1997;18:533–537. - PubMed
-
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. - PubMed
-
- Bader GD, Hogue CW. BIND—A data specification for storing and describing biomolecular interactions, molecular complexes and pathways. Bioinformatics. 2000;16:465–477. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases