

ARACHNE: a whole-genome shotgun assembler
- PMID: 11779843
- PMCID: PMC155255
- DOI: 10.1101/gr.208902
ARACHNE: a whole-genome shotgun assembler
Abstract
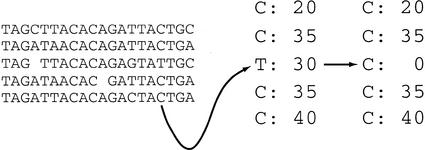
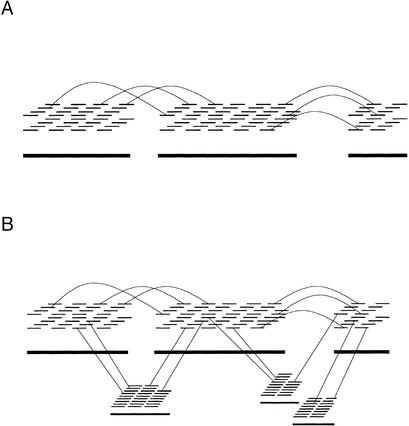
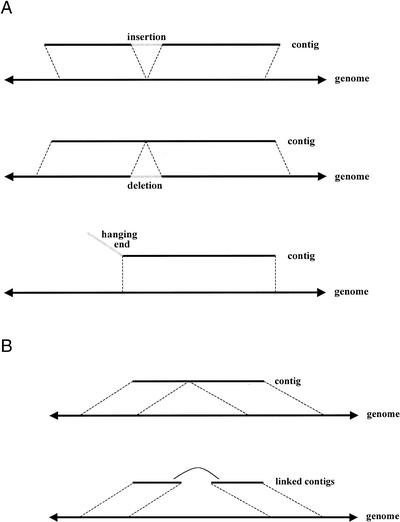
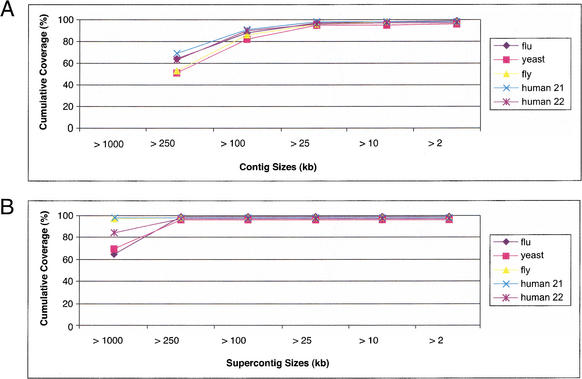
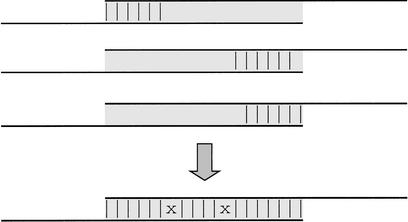
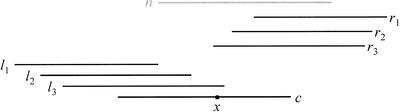
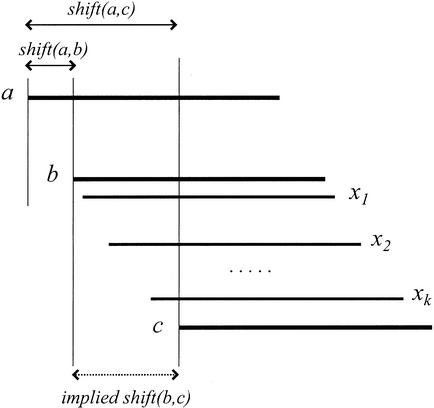
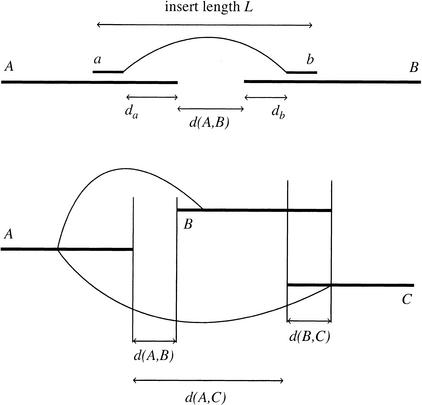
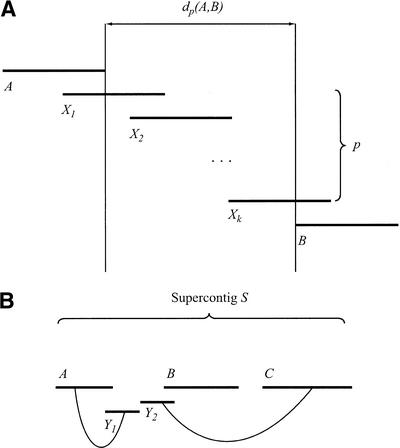
We describe a new computer system, called ARACHNE, for assembling genome sequence using paired-end whole-genome shotgun reads. ARACHNE has several key features, including an efficient and sensitive procedure for finding read overlaps, a procedure for scoring overlaps that achieves high accuracy by correcting errors before assembly, read merger based on forward-reverse links, and detection of repeat contigs by forward-reverse link inconsistency. To test ARACHNE, we created simulated reads providing approximately 10-fold coverage of the genomes of H. influenzae, S. cerevisiae, and D. melanogaster, as well as human chromosomes 21 and 22. The assemblies of these simulated reads yielded nearly complete coverage of the respective genomes, with a small number of contigs joined into a smaller number of supercontigs (or scaffolds). For example, analysis of the D. melanogaster genome yielded approximately 98% coverage with an N50 contig length of 324 kb and an N50 supercontig length of 5143 kb. The assembly accuracy was high, although not perfect: small errors occurred at a frequency of roughly 1 per 1 Mb (typically, deletion of approximately 1 kb in size), with a very small number of other misassemblies. The assembly was rapid: the Drosophila assembly required only 21 hours on a single 667 MHz processor and used 8.4 Gb of memory.
Figures
References
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
-
- Batzoglou S. “Computational genomics: Mapping, comparison, and annotation of genomes.” Ph.D. dissertion. Massachusetts Institute of Technology, Department of Electrical Engineering and Computer Science; 2000.
-
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. - PubMed
-
- Chen, T. and Skiena, S.S. 1997. Trie-based data structures for sequence assembly, In: Proceedings of The Eighth Symposium on Combinatorial Pattern Matching, pp. 206–223. Springer-Verlag, New York.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous