

Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes
- PMID: 11781353
- PMCID: PMC150813
- DOI: 10.1172/JCI11198
Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes
Abstract
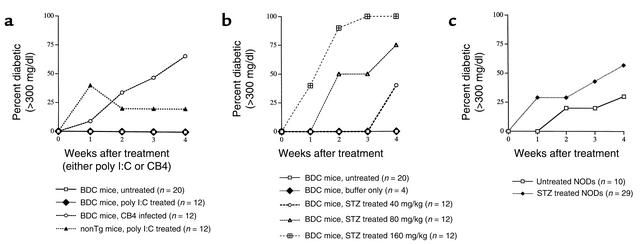
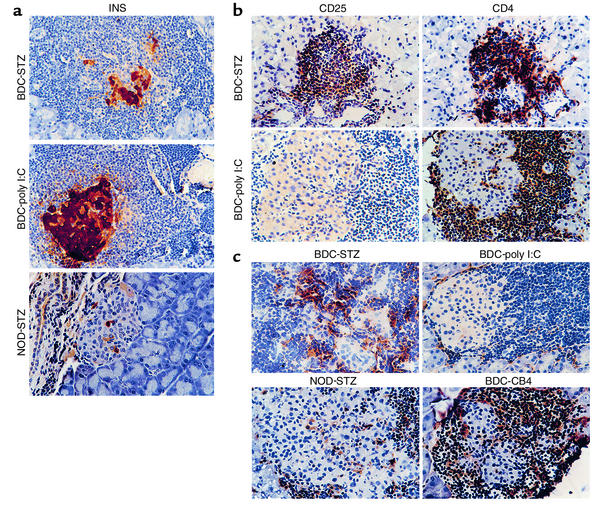
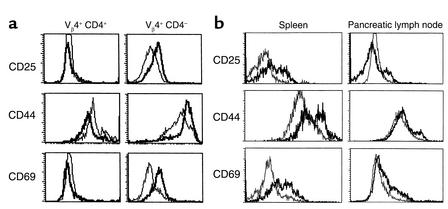
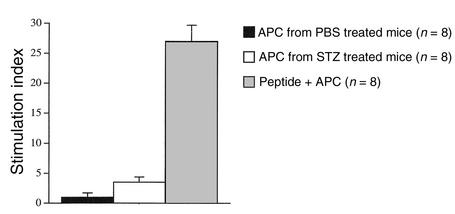
The induction of autoimmunity by viruses has been attributed to numerous mechanisms. In mice, coxsackievirus B4 (CB4) induces insulin-dependent diabetes mellitus (IDDM) resembling the final step of disease progression in humans. The immune response following the viral insult clearly precipitates IDDM. However, the molecular pathway between viral infection and the subsequent activation of T cells specific for islet antigen has not been elucidated. These T cells could become activated through exposure to sequestered antigens released by damaged beta cells, or they could have responded to factors secreted by the inflammatory response itself. To distinguish between these possibilities, we treated mice harboring a diabetogenic T cell repertoire with either the islet-damaging agent streptozotocin (STZ) or poly I:C, which nonspecifically activates T cells. Significantly, only treatment of mice with STZ resulted in IDDM and mimicked the effects observed following CB4 infection. Furthermore, antigen-presenting cells from STZ-treated mice were shown to directly activate autoreactive T cells and induce diabetes. Therefore, the primary role of CB4 in the precipitation of IDDM is to damage tissue, causing release and presentation of sequestered islet antigen. These events stimulate autoreactive T cells and thereby initiate disease.
Figures
Similar articles
-
Requirements for viral-mediated autoimmune diabetes: beta-cell damage and immune infiltration.J Autoimmun. 2001 May;16(3):211-7. doi: 10.1006/jaut.2000.0486. J Autoimmun. 2001. PMID: 11334485
-
Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells.Clin Immunol. 2004 Feb;110(2):134-44. doi: 10.1016/j.clim.2003.09.014. Clin Immunol. 2004. PMID: 15003810
-
Pathological changes in the islet milieu precede infiltration of islets and destruction of beta-cells by autoreactive lymphocytes in a transgenic model of virus-induced IDDM.J Autoimmun. 1997 Jun;10(3):231-8. doi: 10.1006/jaut.1997.0131. J Autoimmun. 1997. PMID: 9218748
-
Autoimmune diabetes: the role of T cells, MHC molecules and autoantigens.Autoimmunity. 1998;27(3):159-77. doi: 10.3109/08916939809003864. Autoimmunity. 1998. PMID: 9609134 Review.
-
Autoimmune disorders in diabetes.Adv Nephrol Necker Hosp. 1986;15:281-305. Adv Nephrol Necker Hosp. 1986. PMID: 3082114 Review.
Cited by
-
Transgenic expression of Hsc70 in pancreatic islets enhances autoimmune diabetes in response to beta cell damage.J Immunol. 2009 Nov 1;183(9):5728-37. doi: 10.4049/jimmunol.0901288. Epub 2009 Oct 7. J Immunol. 2009. PMID: 19812207 Free PMC article.
-
Monkeypox virus and type 1 diabetes: a molecular insight into inflammatory signaling and β-cell autoimmunity.Virol J. 2025 Jun 14;22(1):195. doi: 10.1186/s12985-025-02822-7. Virol J. 2025. PMID: 40517247 Free PMC article. Review.
-
Natural endogenous adjuvants.Springer Semin Immunopathol. 2005 Jan;26(3):231-46. doi: 10.1007/s00281-004-0173-3. Epub 2004 Oct 14. Springer Semin Immunopathol. 2005. PMID: 15609001 Review.
-
Phenylmethimazole suppresses dsRNA-induced cytotoxicity and inflammatory cytokines in murine pancreatic beta cells and blocks viral acceleration of type 1 diabetes in NOD mice.Molecules. 2013 Mar 27;18(4):3841-58. doi: 10.3390/molecules18043841. Molecules. 2013. PMID: 23535518 Free PMC article.
-
Autoimmune disease triggered by infection with alphaproteobacteria.Expert Rev Clin Immunol. 2009 Jul 1;5(4):369-379. doi: 10.1586/ECI.09.23. Expert Rev Clin Immunol. 2009. PMID: 20161124 Free PMC article.
References
-
- Andreoletti L, et al. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J Med Virol. 1997; 52:121–127. - PubMed
-
- Hyoty H, et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes. 1995; 44:652–657. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
