

Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis
- PMID: 11781872
- PMCID: PMC384921
- DOI: 10.1086/338758
Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis
Abstract
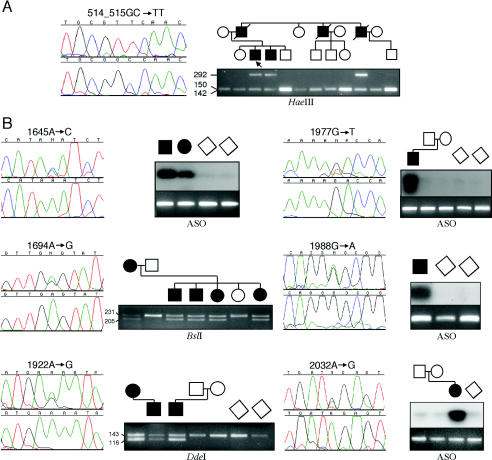
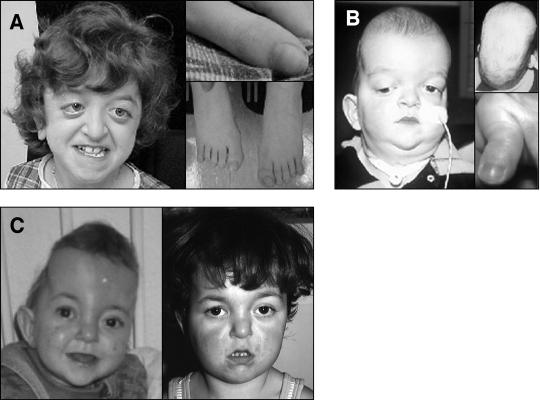
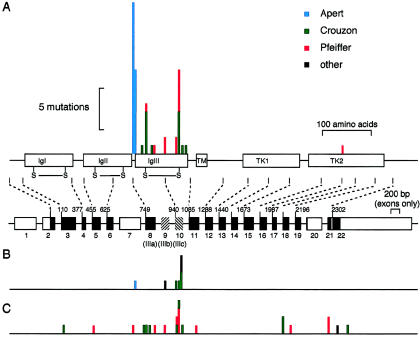
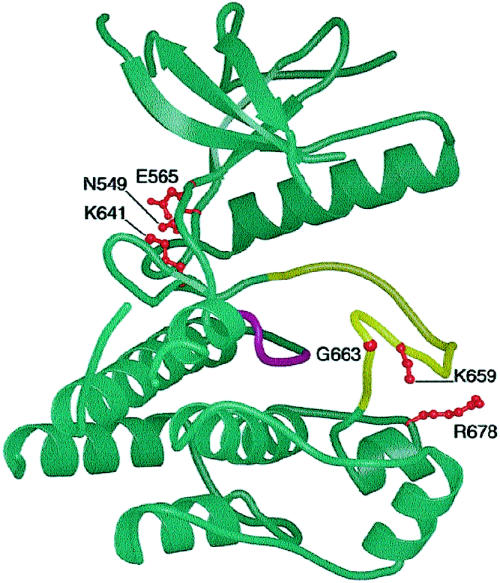
It has been known for several years that heterozygous mutations of three members of the fibroblast growth-factor-receptor family of signal-transduction molecules-namely, FGFR1, FGFR2, and FGFR3-contribute significantly to disorders of bone patterning and growth. FGFR3 mutations, which predominantly cause short-limbed bone dysplasia, occur in all three major regions (i.e., extracellular, transmembrane, and intracellular) of the protein. By contrast, most mutations described in FGFR2 localize to just two exons (IIIa and IIIc), encoding the IgIII domain in the extracellular region, resulting in syndromic craniosynostosis including Apert, Crouzon, or Pfeiffer syndromes. Interpretation of this apparent clustering of mutations in FGFR2 has been hampered by the absence of any complete FGFR2-mutation screen. We have now undertaken such a screen in 259 patients with craniosynostosis in whom mutations in other genes (e.g., FGFR1, FGFR3, and TWIST) had been excluded; part of this screen was a cohort-based study, enabling unbiased estimates of the mutation distribution to be obtained. Although the majority (61/62 in the cohort sample) of FGFR2 mutations localized to the IIIa and IIIc exons, we identified mutations in seven additional exons-including six distinct mutations of the tyrosine kinase region and a single mutation of the IgII domain. The majority of patients with atypical mutations had diagnoses of Pfeiffer syndrome or Crouzon syndrome. Overall, FGFR2 mutations were present in 9.8% of patients with craniosynostosis who were included in a prospectively ascertained sample, but no mutations were found in association with isolated fusion of the metopic or sagittal sutures. We conclude that the spectrum of FGFR2 mutations causing craniosynostosis is wider than previously recognized but that, nevertheless, the IgIIIa/IIIc region represents a genuine mutation hotspot.
Figures

References
Electronic-Database Information
-
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for FGFR2 [accession numbers AC009988, AF410480, AF360695, and NM_000141])
-
- Human Gene Mutation Database, http://archive.uwcm.ac.uk/uwcm/mg/hgmd/search.html (for mutations in KIT, RET, and MET)
-
- KinMutBase protein-alignment web site, http://protein.uta.fi/KinMutBase/ProtAlign.html (for mutations in kinase domains)
-
- Online Mendelian Inheritance in Man (OMIM), http://www3.ncbi.nlm.nih.gov/Omim/searchomim.html (for AS [MIM 101200], CS [MIM 123500], PS [MIM 101600], Beare-Stevenson cutis gyrata syndrome [MIM 123790], and FGFR2 [MIM 176943])
-
- Single Nucleotide Polymorphism web site, http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=2263 (for single-nucleotide polymorphisms in FGFR2)
References
-
- Anderson J, Burns HD, Enriquez-Harris P, Wilkie AOM, Heath JK (1998) Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum Mol Genet 7:1475–1483 - PubMed
-
- Bateman A, Chothia C (1995) Outline structures for the extracellular domains of the fibroblast growth factor receptors. Nat Struct Biol 2:1068–1074 - PubMed
-
- Bellus GA, Gaudenz K, Zackai EH, Clarke LA, Szabo J, Francomano CA, Muenke M (1996) Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet 14:174–176 - PubMed
-
- Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA (2000) Distinct missense mutations of the FGFR3 Lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am J Hum Genet 67:1411–1421 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
