

A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis
- PMID: 11788711
- PMCID: PMC99818
- DOI: 10.1093/nar/30.2.482
A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis
Abstract
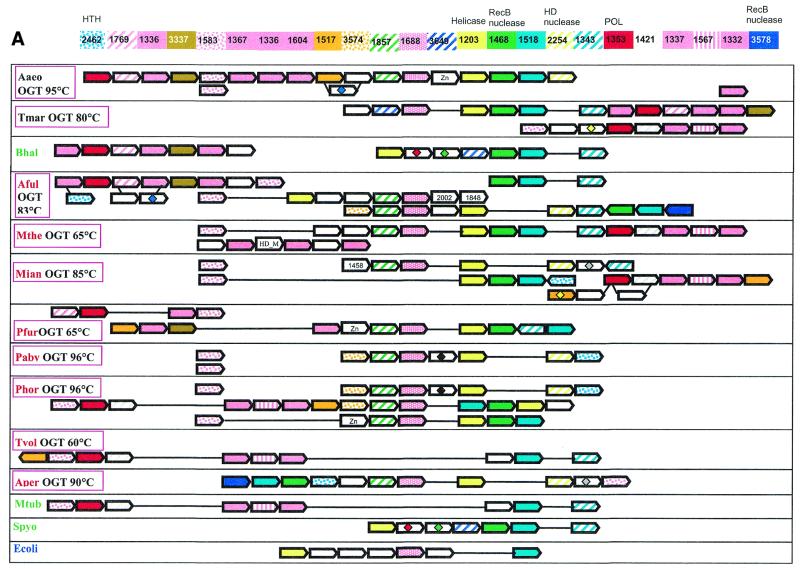
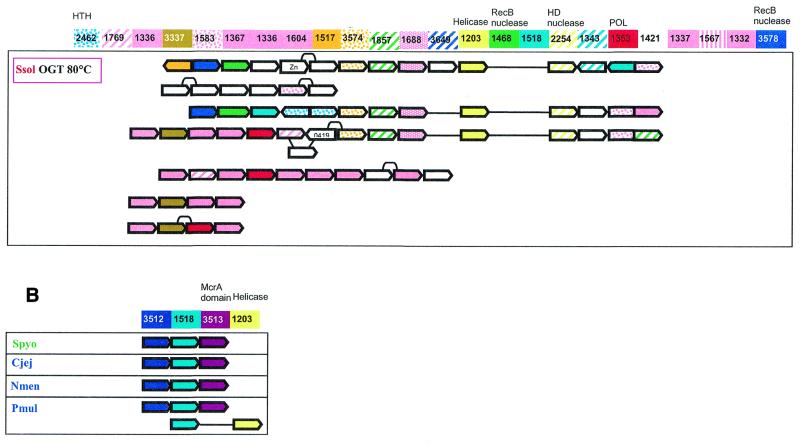
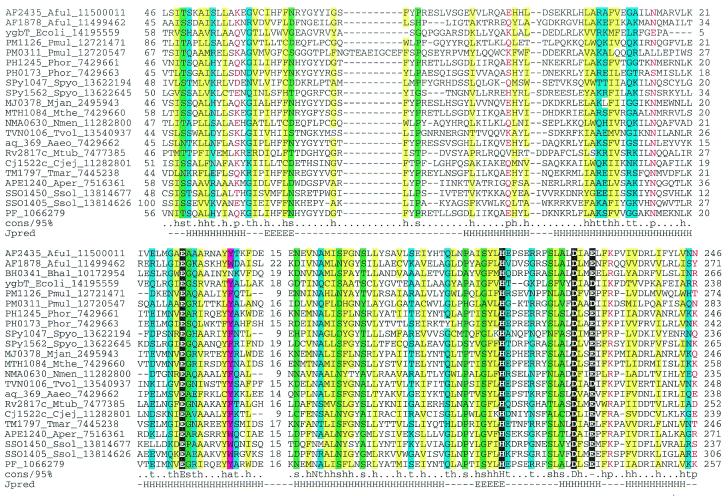
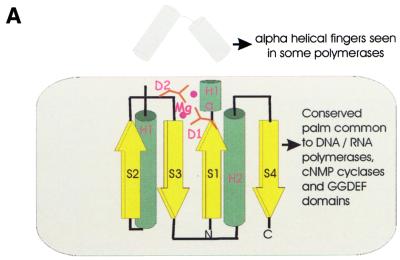
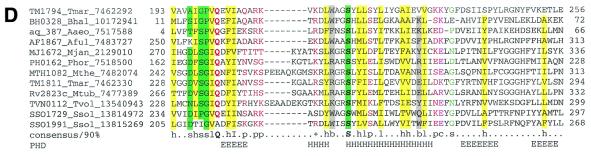
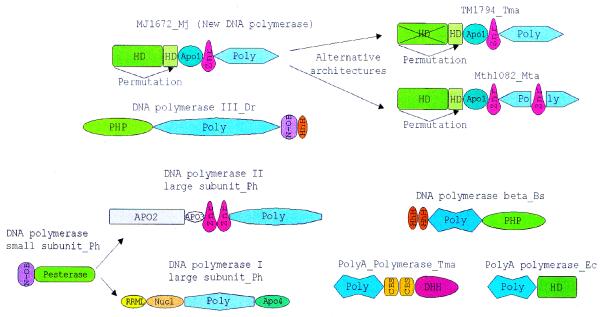
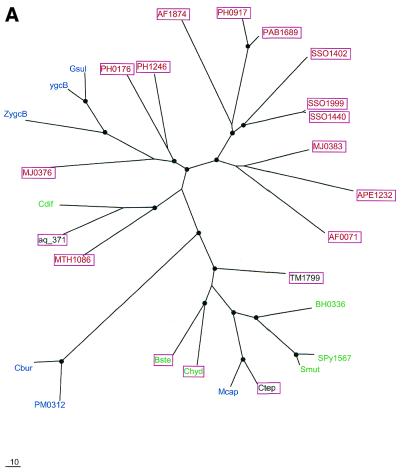
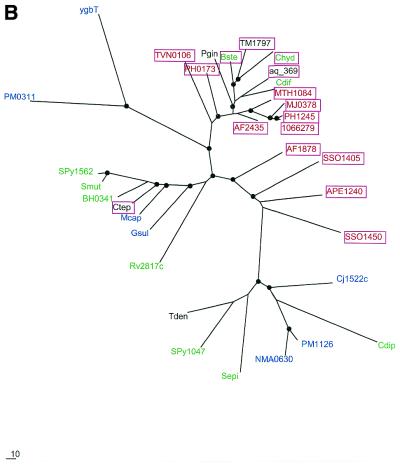
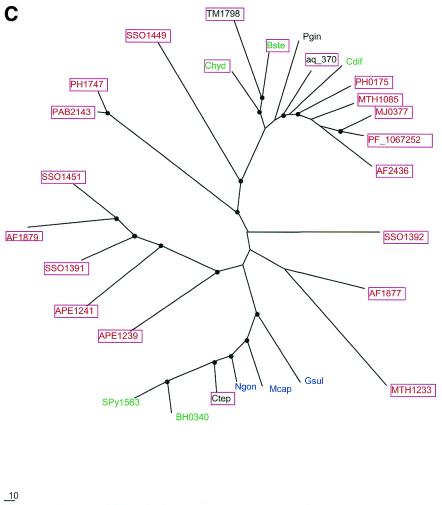
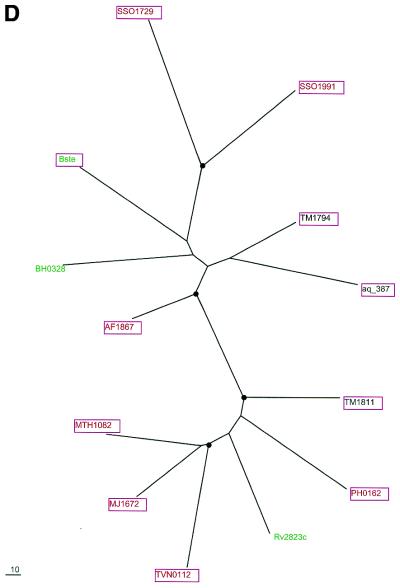
During a systematic analysis of conserved gene context in prokaryotic genomes, a previously undetected, complex, partially conserved neighborhood consisting of more than 20 genes was discovered in most Archaea (with the exception of Thermoplasma acidophilum and Halobacterium NRC-1) and some bacteria, including the hyperthermophiles Thermotoga maritima and Aquifex aeolicus. The gene composition and gene order in this neighborhood vary greatly between species, but all versions have a stable, conserved core that consists of five genes. One of the core genes encodes a predicted DNA helicase, often fused to a predicted HD-superfamily hydrolase, and another encodes a RecB family exonuclease; three core genes remain uncharacterized, but one of these might encode a nuclease of a new family. Two more genes that belong to this neighborhood and are present in most of the genomes in which the neighborhood was detected encode, respectively, a predicted HD-superfamily hydrolase (possibly a nuclease) of a distinct family and a predicted, novel DNA polymerase. Another characteristic feature of this neighborhood is the expansion of a superfamily of paralogous, uncharacterized proteins, which are encoded by at least 20-30% of the genes in the neighborhood. The functional features of the proteins encoded in this neighborhood suggest that they comprise a previously undetected DNA repair system, which, to our knowledge, is the first repair system largely specific for thermophiles to be identified. This hypothetical repair system might be functionally analogous to the bacterial-eukaryotic system of translesion, mutagenic repair whose central components are DNA polymerases of the UmuC-DinB-Rad30-Rev1 superfamily, which typically are missing in thermophiles.
Figures





References
-
- Stetter K.O. (1996) Hyperthermophiles in the history of life. Ciba Found Symp., 202, 1–10. - PubMed
-
- Nisbet E. (2000) The realms of Archaean life. Nature, 405, 625–626. - PubMed
-
- Grogan D.W. (2000) The question of DNA repair in hyperthermophilic archaea. Trends Microbiol., 8, 180–185. - PubMed
-
- Watrin L. and Prieur,D. (1996) UV and ethyl methanesulfonate effects in hyperthermophilic archaea and isolation of auxotrophic mutants of Pyrococcus strains. Curr. Microbiol., 33, 377–382. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
