

Convergence and constraint in eukaryotic release factor 1 (eRF1) domain 1: the evolution of stop codon specificity
- PMID: 11788716
- PMCID: PMC99827
- DOI: 10.1093/nar/30.2.532
Convergence and constraint in eukaryotic release factor 1 (eRF1) domain 1: the evolution of stop codon specificity
Abstract
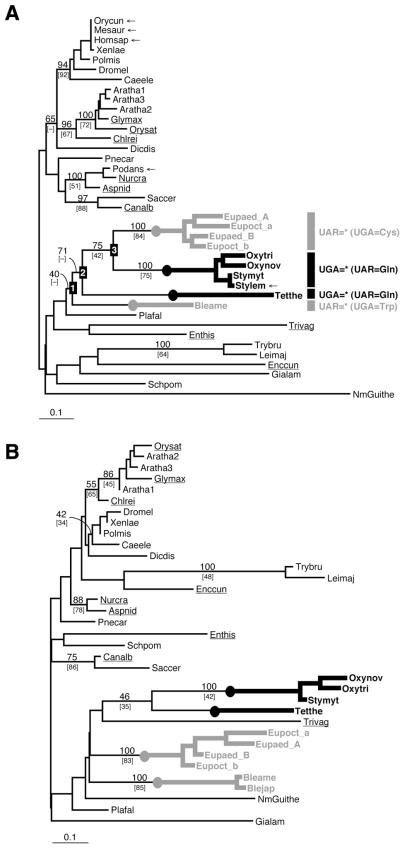
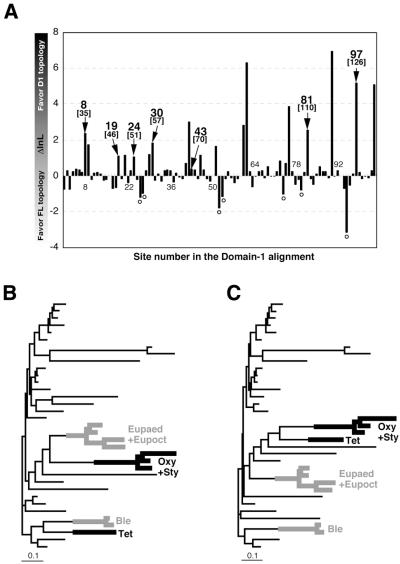
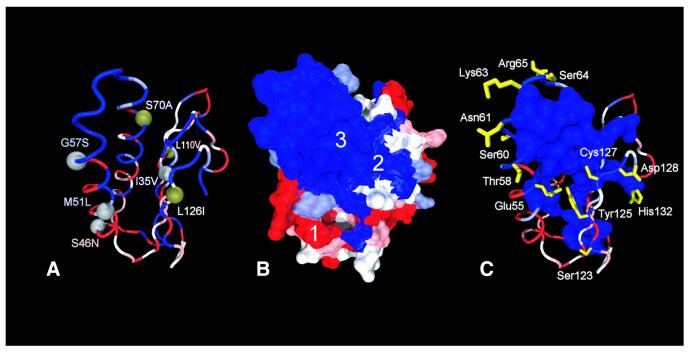
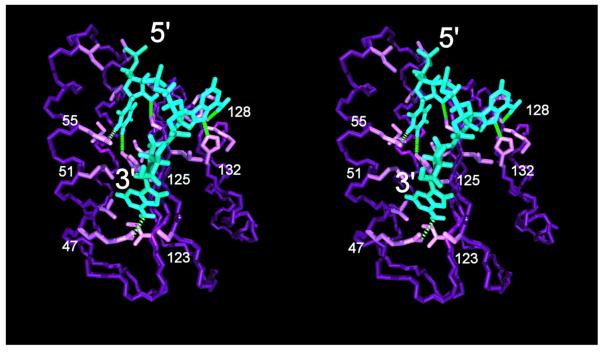
Class 1 release factor in eukaryotes (eRF1) recognizes stop codons and promotes peptide release from the ribosome. The 'molecular mimicry' hypothesis suggests that domain 1 of eRF1 is analogous to the tRNA anticodon stem-loop. Recent studies strongly support this hypothesis and several models for specific interactions between stop codons and residues in domain 1 have been proposed. In this study we have sequenced and identified novel eRF1 sequences across a wide diversity of eukaryotes and re-evaluated the codon-binding site by bioinformatic analyses of a large eRF1 dataset. Analyses of the eRF1 structure combined with estimates of evolutionary rates at amino acid sites allow us to define the residues that are under structural (i.e. those involved in intramolecular interactions) versus non-structural selective constraints. Furthermore, we have re-assessed convergent substitutions in the ciliate variant code eRF1s using maximum likelihood-based phylogenetic approaches. Our results favor the model proposed by Bertram et al. that stop codons bind to three 'cavities' on the protein surface, although we suggest that the stop codon may bind in the opposite orientation to the original model. We assess the feasibility of this alternative binding orientation with a triplet stop codon and the eRF1 domain 1 structures using molecular modeling techniques.
Figures

References
-
- Nakamura Y. and Ito,K. (1998) How proteins read the stop codon and terminate translation. Genes Cells, 3, 265–278. - PubMed
-
- Kisselev L.L. and Buckingham,R.H. (2000) Translational termination comes of age. Trends Biochem. Sci., 25, 561–566. - PubMed
-
- Poole E. and Tate,W. (2000) Release factors and their role as decoding proteins: specificity and fidelity for termination of protein synthesis. Biochim. Biophys. Acta, 1493, 1–11. - PubMed
-
- Bertram G., Innes,S., Minella,O., Richardson,J. and Stansfield,I. (2001) Endless possibilities: translation termination and stop codon recognition. Microbiology, 147, 255–269. - PubMed
-
- Dontsova M., Frolova,L., Vassilieva,J., Piendl,W., Kisselev,L. and Garber,M. (2000) Translation termination factor aRF1 from the archaeon Methanococcus jannaschii is active with eukaryotic ribosomes. FEBS Lett., 472, 213–216. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
