

Quod erat demonstrandum? The mystery of experimental validation of apparently erroneous computational analyses of protein sequences
- PMID: 11790254
- PMCID: PMC64836
- DOI: 10.1186/gb-2001-2-12-research0051
Quod erat demonstrandum? The mystery of experimental validation of apparently erroneous computational analyses of protein sequences
Abstract
Background: Computational predictions are critical for directing the experimental study of protein functions. Therefore it is paradoxical when an apparently erroneous computational prediction seems to be supported by experiment.
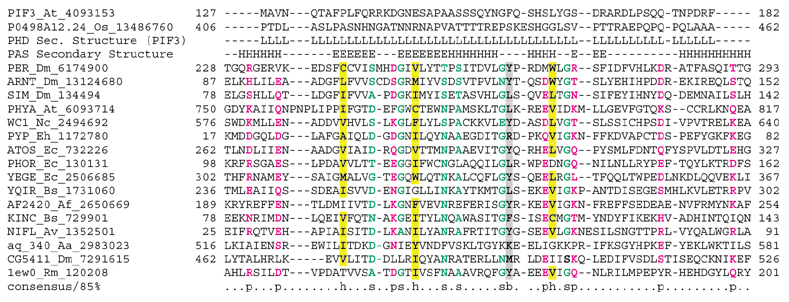
Results: We analyzed six cases where application of novel or conventional computational methods for protein sequence and structure analysis led to non-trivial predictions that were subsequently supported by direct experiments. We show that, on all six occasions, the original prediction was unjustified, and in at least three cases, an alternative, well-supported computational prediction, incompatible with the original one, could be derived. The most unusual cases involved the identification of an archaeal cysteinyl-tRNA synthetase, a dihydropteroate synthase and a thymidylate synthase, for which experimental verifications of apparently erroneous computational predictions were reported. Using sequence-profile analysis, multiple alignment and secondary-structure prediction, we have identified the unique archaeal 'cysteinyl-tRNA synthetase' as a homolog of extracellular polygalactosaminidases, and the 'dihydropteroate synthase' as a member of the beta-lactamase-like superfamily of metal-dependent hydrolases.
Conclusions: In each of the analyzed cases, the original computational predictions could be refuted and, in some instances, alternative strongly supported predictions were obtained. The nature of the experimental evidence that appears to support these predictions remains an open question. Some of these experiments might signify discovery of extremely unusual forms of the respective enzymes, whereas the results of others could be due to artifacts.
Figures







References
-
- Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y. Predicting function: from genes to genomes and back. J Mol Biol. 1998;283:707–725. - PubMed
-
- Koonin EV, Aravind L, Kondrashov AS. The impact of comparative genomics on our understanding of evolution. Cell. 2000;101:573–576. - PubMed
-
- Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J Mol Biol. 1999;287:1023–1040. - PubMed
-
- Murzin AG. Progress in protein structure prediction. Nat Struct Biol. 2001;8:110–112. - PubMed
-
- Karlin S, Bucher P, Brendel V, Altschul SF. Statistical methods and insights for protein and DNA sequences. Annu Rev Biophys Biophys Chem. 1991;20:175–203. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
