

Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations
- PMID: 11790832
- PMCID: PMC2373438
- DOI: 10.1110/ps.33202
Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations
Abstract
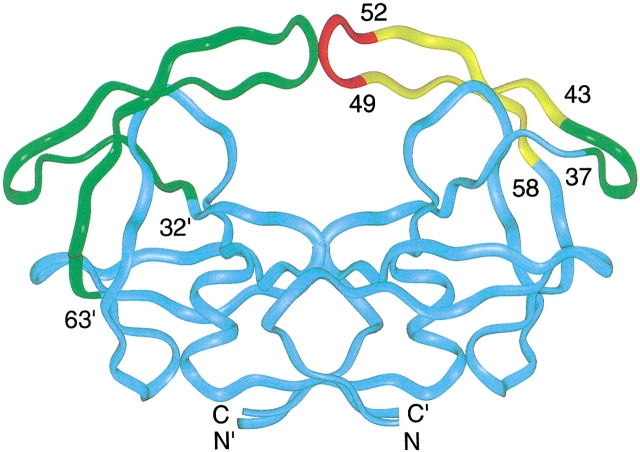
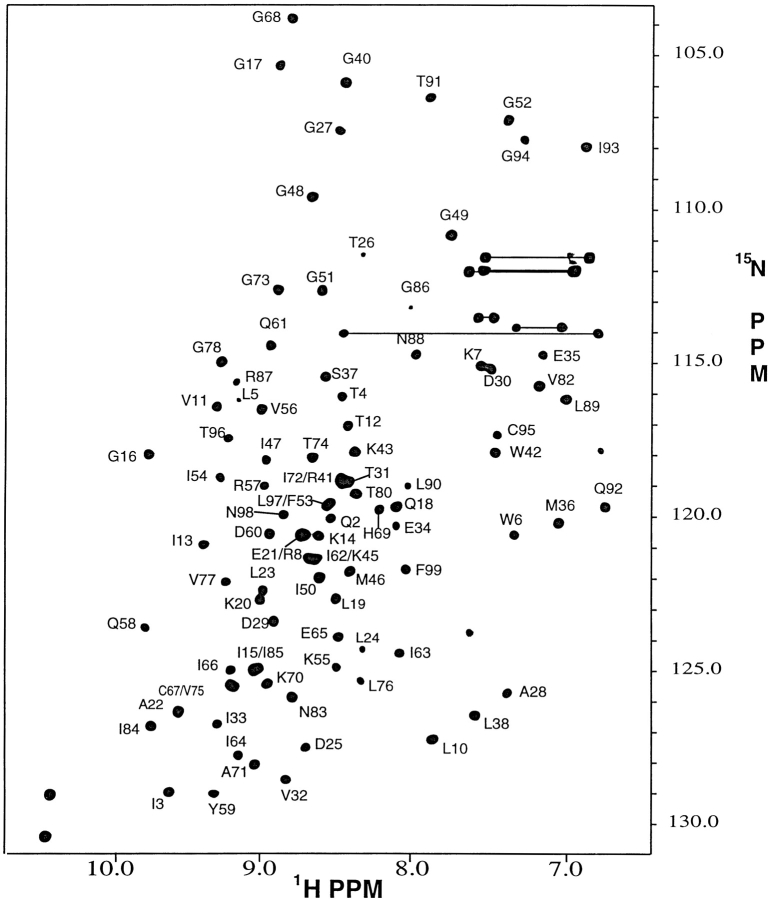
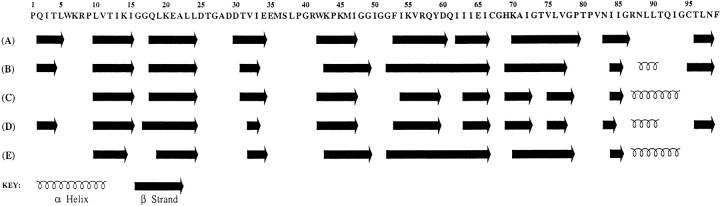
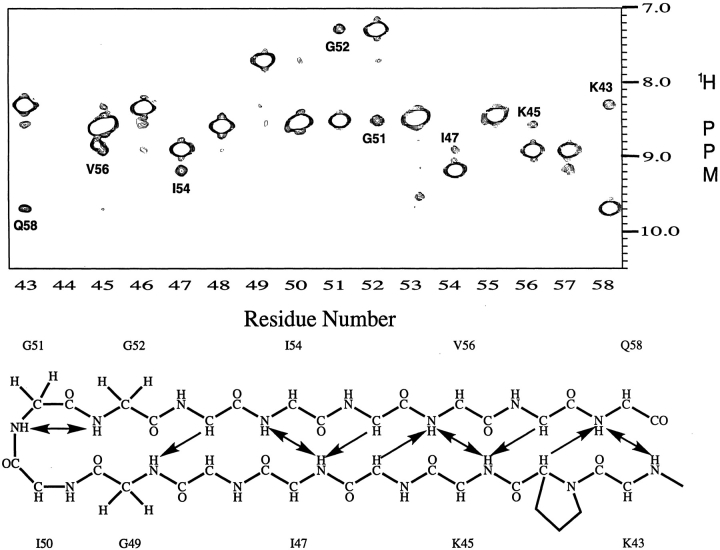
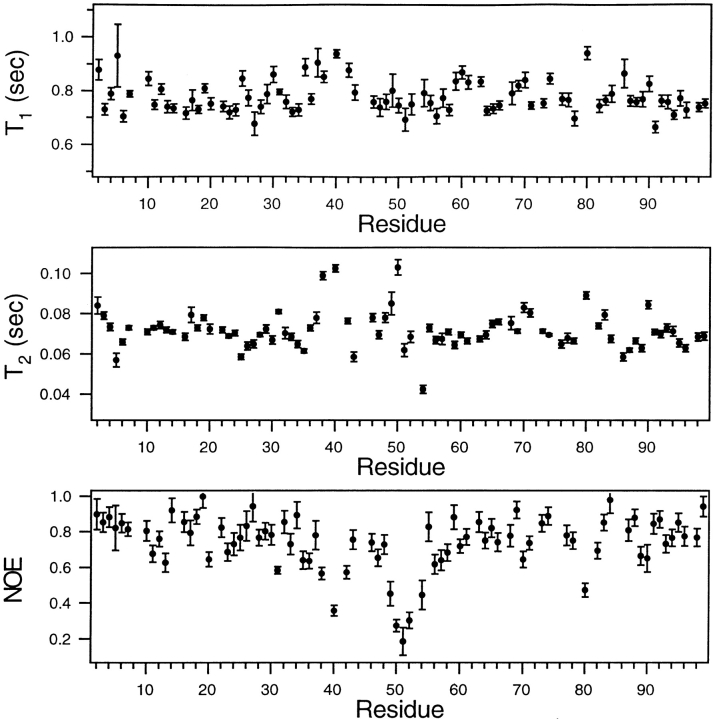
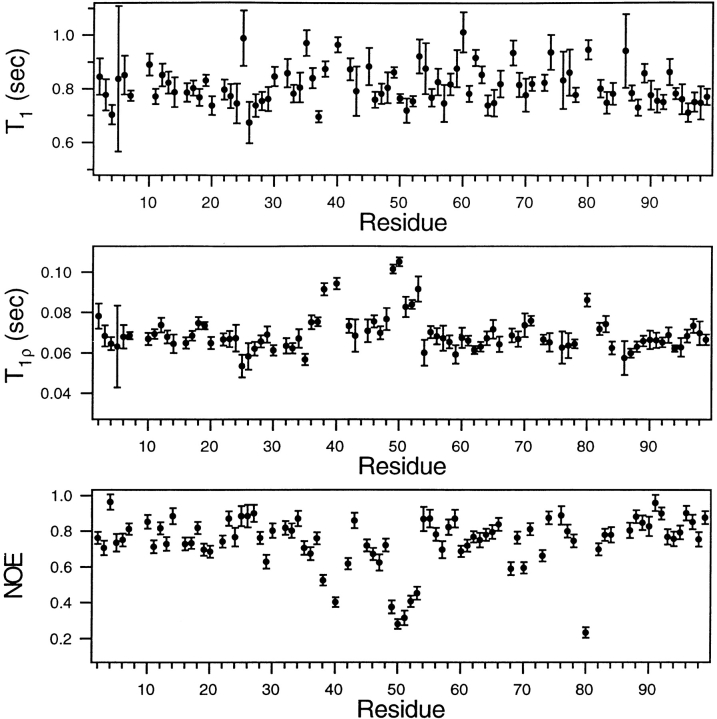
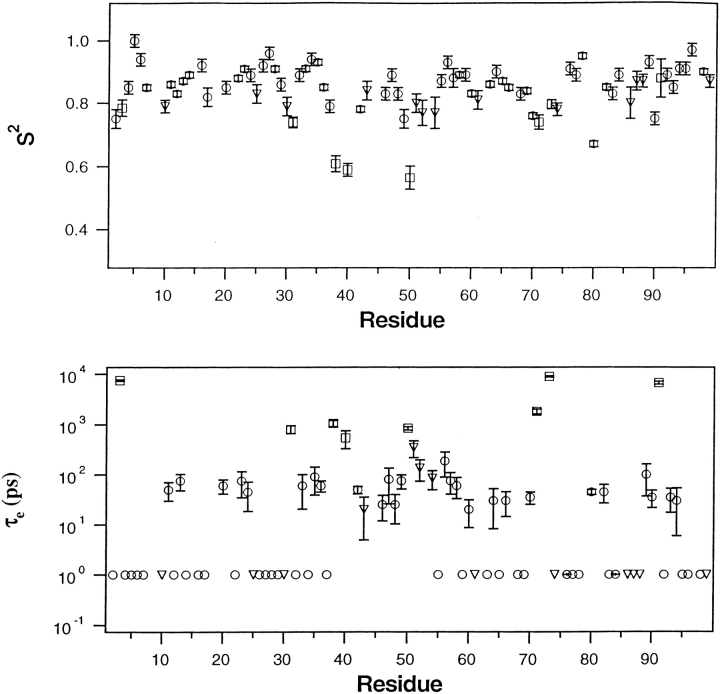
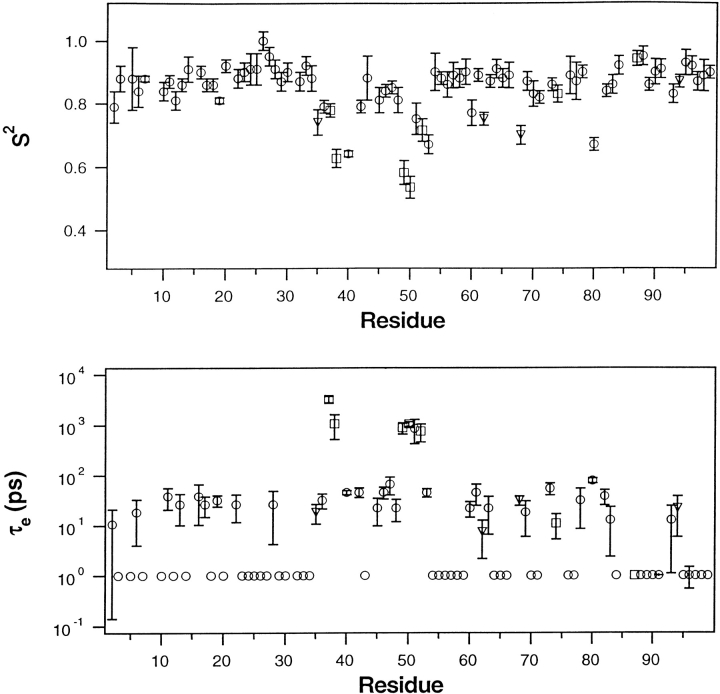
Crystal structures have shown that the HIV-1 protease flaps, domains that control access to the active site, are closed when the active site is occupied by a ligand. Although flap structures ranging from closed to semi-open are observed in the free protease, crystal structures reveal that even the semi-open flaps block access to the active site, indicating that the flaps are mobile in solution. The goals of this paper are to characterize the secondary structure and fast (sub-ns) dynamics of the flaps of the free protease in solution, to relate these results to X-ray structures and to compare them with predictions of dynamics calculations. To this end we have obtained nearly complete backbone and many sidechain signal assignments of a fully active free-protease construct that is stabilized against autoproteolysis by three point mutations. The secondary structure of this protein was characterized using the chemical shift index, measurements of (3h)J(NC') couplings across hydrogen bonds, and NOESY connectivities. Analysis of these measurements indicates that the protease secondary structure becomes irregular near the flap tips, residues 49-53. Model-free analysis of (15)N relaxation parameters, T(1), T(2) (T(1rho)) and (15)N-[(1)H] NOE, shows that residues in the flap tips are flexible on the sub-ns time scale, in contrast with previous observations on the inhibitor-bound protease. These results are compared with theoretical predictions of flap dynamics and the possible biological significance of the sub-ns time scale dynamics of the flap tips is discussed.
Figures
References
-
- Bruschweiler, R., Liao, X.B., and Wright, P.E. 1995. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268: 886–889. - PubMed
-
- Clore, G.M., Szabo, A., Bax, A., Kay, L.E., Driscoll, P.C., and Gronenborn, A. 1990. Deviation from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 112: 4989–4991.
-
- Collins, J.R., Burt, S.K., and Erickson, J.W. 1995. Flap opening in HIV-1 protease simulated by `activated' molecular dynamics. Nat. Struct. Biol. 2: 334–338. - PubMed
-
- Cordier, F. and Grzesiek, S. 1999. Direct observation of hydrogen bonds in proteins by interresidue 3hJNC` scalar couplings. J. Am. Chem. Soc. 121: 1601–1602. - PubMed
-
- Cornilescu, G., Ramirez, B.E., Frank, M.K., Clore, G.M., Gronenborn, A.M., and Bax, A. 1999. Correlation between 3hJNC` and hydrogen bond length in proteins. J. Am. Chem. Soc. 121: 6275–6279.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
